
{kind=link}
There is a tension inherent in the drug approval process between the desire to approve new drugs rapidly in order to treat suffering people and the need to be cautious, to make sure that new drugs are safe and effective before they are approved for sale. This weighing of the risks of too-rapid approval of drugs that doesn’t work (or doesn’t work well) and might cause harm versus the harm that can be caused by delaying approval of an effective drug that might help millions longer than necessary is complex, as is balancing the benefits of rapidly approving effective drugs versus the benefits of a more rigorous approval process that catches potentially dangerous drugs before they are approved.
Even under ideal circumstances, value judgments must be made, but add politics and profit to the mix, and it’s a very contentious process, with multiple players, most of whom don’t like delay. Drug companies want their drugs approved rapidly because of the potential profit. Patient advocates with serious diseases want promising new drugs sooner. Conservatives opposed to regulation like to push for faster drug approval as a means of making the Food and Drug Administration (FDA) less powerful, which was, of course, the origin of unethical “right-to-try” laws. Those harboring distrust of big pharma (which is often justified) want a more rigorous approval because they believe that the existing process is rigged in favor of the drug companies and don’t trust its results. It’s an uneasy tension that’s existed for many decades, ever since the formation of the FDA in 1906, through the 1962 Kefauver-Harris Drug Amendments to the Federal Food, Drug, and Cosmetic Act, which mandated evidence of efficacy in addition to evidence of safety before a drug could be approved. This was a law passed in the wake of the thalidomide disaster.
Since then, depending on who was in office and who the FDA Commissioner was, the pendulum has shifted back and forth over how rigorously safety and efficacy have to be show before a drug can be approved. In response to the HIV/AIDS crisis, for instance, the accelerated approval process was initiated to allow approval for drugs that fill an urgent unmet need for the treatment of serious conditions to be approved based on surrogate endpoints:
The FDA instituted its Accelerated Approval Program to allow for earlier approval of drugs that treat serious conditions, and that fill an unmet medical need based on a surrogate endpoint. A surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval.
Drug companies are still required to conduct studies to confirm the anticipated clinical benefit. These studies are known as phase 4 confirmatory trials. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug. If the confirmatory trial does not show that the drug provides clinical benefit, FDA has regulatory procedures in place that could lead to removing the drug from the market.
It was a reasonable response to a tough problem; or at least it seemed so at the time. Drugs in this pathway could, after 1992, be approved by the FDA if their makers could demonstrate an effect on a surrogate measure or intermediate clinical end point that is, according to the program, “reasonably likely” to predict a real clinical end point, such as changes in symptoms or mortality rates. Certainly, surrogate endpoints can be measured in a much shorter time frame than the several years it usually takes to identify improvements in overall survival. The accelerated approval program, however, rests on a big assumption, namely that the surrogate endpoint chosen is a good predictor of clinical outcome. So it’s reasonable to ask: How often do drugs approved through this process ultimately pan out and provide significant benefit to patients? A study asking this question was published in JAMA Internal Medicine last week, which made it into the news:
Many cancer patients and oncologists may turn to the US Food and Drug Administration’s accelerated approval program, which provides access to new cancer drugs faster — especially when a life-threatening cancer needs swift treatment or relief.
Yet a new study questions the clinical benefit of some drugs in the program and suggests ways in which the program could be improved.
It turns out that the study, carried out by researchers at Brigham and Women’s Hospital and Queen’s University in Canada, found that only one-fifth of oncology drugs that received accelerated approval were ultimately validated in confirmatory trials to have an effect on overall survival. Basically, the study calls into question the entire rationale of the FDA’s accelerated approval system, at least in oncology. Let’s dig in.
Surrogate endpoints in oncology versus real endpoints
Cancer therapies are generally evaluated using a number of endpoints. The most commonly used include overall survival (OS), progression-free survival (PFS), or recurrence-free survival (RFS). There are, of course, other endpoints related to survival that are measured, but these three are the most relevant for this discussion. Overall survival is what it sounds like: How long do patients survive their cancer after diagnosis? Period. It’s hard for an endpoint to be more objective than that: At a certain timepoint after diagnosis, either the patient is alive or she is dead. This number is usually expressed in terms of median survival, which is the period of time after which half of the patients under study are still alive and half have died. It should be noted that OS includes all causes of death, not just cancer. If a patient with cancer under study dies of a heart attack that is not related to his cancer or his cancer treatment, that counts.
Traditionally, OS has been the “gold standard” endpoint in measuring the efficacy of a cancer therapy, because the primary goal has been to prolong survival, the ideal case being prolonging survival to the point where it is indistinguishable from life expectancy if the patient never had cancer in the first place. PFS, on the other hand, is survival without progression; i.e., how long the patient with cancer survives without evidence that his or her tumor has measurably grown or metastasized, with dying of other causes with disease counting in the calculation. While PFS is often measured as well as OS, it’s generally considered less useful because it is entirely possible for a treatment to prolong PFS without prolonging OS. This sort of result can happen when the treatment is effective at shrinking a tumor and/or slowing its progression but its toxicities contribute to death from other causes. In such a case, PFS improves with no improvement in OS. Finally, RFS (also called disease-free survival, or DFS) is how long the patient survives without a recurrence of her cancer after successful initial treatment. PFS and RFS are often used as surrogate endpoints in cancer therapy clinical trials, although increasingly there is one other. In neoadjuvant chemotherapy (chemotherapy given before surgery), a pathologic complete response (pCR) has traditionally been considered a good surrogate endpoint. A pCR occurs when, at surgery, there are viable cancer cells detectable in the specimen removed at surgery. There is some controversy about this, in breast cancer at least, but in aggressive forms of breast cancer the correlation between pCR and improved overall survival is good, with a relatively recent meta-analysis having found that there is an 84% decreased chance of death in women with an aggressive subtype of cancer known as “triple negative” whose neoadjuvant treatment achieves a pCR.
Other surrogate endpoints include response rate (RR), defined as the percentage of patients whose tumor shrinks significantly in response to the therapy (usually 30% or more is the cutoff); and time to tumor progression (TTP, which is the same as PFS but doesn’t include deaths; it just measures time until the tumor detectably progresses). To these is being added a dizzying array of molecular surrogate endpoints that are less accepted, and it’s hard not to be tempted to pick your favorites if you’re an oncology researcher.
The debate about the use of surrogate endpoints in oncology has been going on as long as I can remember, dating back to before I entered the field of surgical oncology as a fellow back in the mid-1990s, and, as this study shows, is still going on. Unfortunately, the relationship between these surrogate endpoints and overall survival is not always clear or as tight as we would like to think. Nine years ago, I discussed in depth one such case, Avastin (bevacizumab), an angiogenesis inhibitor, and its use for advanced breast cancer. The drug had achieved accelerated approval for this indication in 2008 on the basis of trials showing improvements in PFS. However, by 2010, followup trials had failed to show an improvement in OS, and the FDA rescinded its provisional approval. When Avastin was approved, a prominent breast cancer oncologist justified Avastin’s approval based on PFS thusly:
Dr. Kathy Albain, a breast cancer specialist at Loyola University Medical Center in Maywood, Ill., polled colleagues and patients and found overwhelming support for approving drugs based on delaying tumor progression. It would be ideal to show that a drug also prolongs life, but that may not be realistic, she said. The reason is that when a woman’s cancer progresses, doctors change the drugs they use, hoping to slow the cancer. That dilutes any impact of the first drug — in this case Avastin.
At the time, I noted that, although I could see where Dr. Albain was coming from, I had always been uneasy with this view. I could see the utility of a drug that slows tumor progression but does not prolong survival, but I could only see its usefulness in one situation: There must also be good evidence that that drug also improves quality of life, evidence that was definitely lacking for Avastin, which, if anything, increased complication rates and was very expensive, to boot.
In any event, the FDA’s removal of its approval for Avastin for advanced breast cancer shows another problem with accelerated approval. When a drug is provisionally approved and then its approval rescinded based on later clinical trials, no one is happy. Certainly breast cancer advocates were very unhappy. I even experienced some of their wrath when I foolishly agreed to be on a radio show to discuss the issue. What I wasn’t told was that there would also be a breast cancer advocate on the show as well, an advocate who was receiving Avastin and believed that it was responsible for her continued survival. Politics also intruded. The Affordable Care Act had just been passed into law at the time, and conservative opponents of the ACA attacked the FDA’s decision on Avastin as “evidence” of the impending arrival of socialized medicine-style rationing, thanks, of course, to “Obamacare”, characterizing the decision as the FDA being made complicit in “rationing” health care.
It is a very thorny issue, which is why the study under discussion, by Bishal Gyawali at Brigham and Women’s Hospital, asks a very pertinent question: How often do oncology drugs approved by the accelerated approval mechanism show improvements in OS in the subsequent confirmatory clinical trials required by the FDA?
Disappointing results
To answer this question, Gyawali et al looked at drugs approved by the FDA based on surrogate endpoints. In the introduction, it is noted:
The FDA recently published an article on the 25-year experience with the accelerated approval pathway that examined the fate of 93 oncology indications granted accelerated approval from December 11, 1992, through May 31, 2017.9 The review found that 81 (87%) of the original 93 accelerated approvals were based on response rates—a surrogate marker in which the effect of an intervention is determined based on a change in tumor size. In addition, 8 (9%) of 93 accelerated approvals were based on PFS or time to tumor progression (TTP) and 4 (4%) were based on DFS or recurrence-free survival (RFS). Progression-free survival and TTP are surrogate markers that measure the time between the start of treatment and tumor growth beyond a certain size in the case of metastatic disease, whereas DFS and RFS measure the time from the start of treatment to disease recurrence when the drug is used as adjuvant therapy.
The FDA reported that in 51 (55%) of the 93 indications, confirmatory trials verified clinical benefit. In 5 cases (5%), approval for an indication was withdrawn in light of postapproval trial results, and postapproval evaluations were ongoing for the remaining 37 (40%) indications.9 The FDA concluded that the low failure rate in confirmatory trials was evidence that the accelerated approval pathway was operating effectively. We assessed the nature of the end points used for the verification of benefit in confirmatory trials and provide an update on the current status of the remaining indications for which confirmatory trials were ongoing at the time of the FDA’s analysis.
Gyawali et al wanted to update the outcomes reported by the FDA. So in May 2018, which was one year after closure of data collection for the FDA’s study, they searched PubMed, as well as the FDA database of postmarketing requirements and commitments, to determine the current status for postmarket trials for the indications that were labeled as “ongoing” in the FDA report. Confirmatory trials were categorized into three groups: (1) a trial that used OS or a quality-of-life end point, (2) a trial that used a surrogate measure different from the one used in the preapproval trial, and (3) a trial that used the same surrogate measure used in the preapproval trial. Here is the “money table,” which summarizes the study’s findings:
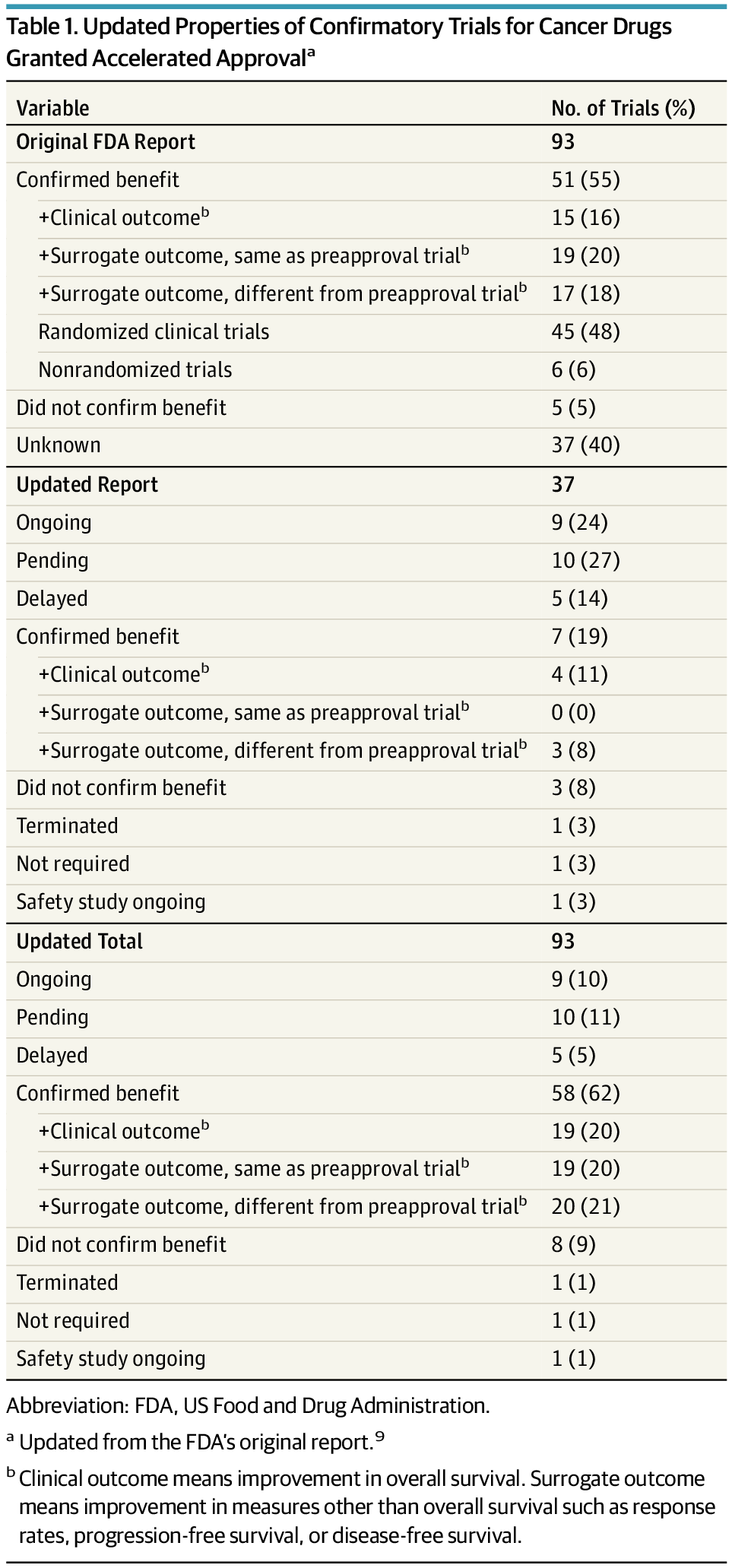
So, looking at the updated figures, I see that on the surface, it looks as though, for 62% of oncology drug approvals, clinical benefit was confirmed. But if you look more closely, you’ll see that only 20% of all 93 cancer drug accelerated approvals, or one-third of the ones claiming confirmation, had improvement in OS noted in confirmatory trials. What about the other 40%? 20% had improvement in the same surrogate in their confirmatory trial, which led me to a major facepalm when I read this result. What on earth is the purpose of just doing another study using the same surrogate endpoint? How does that “confirm” anything? The other 21% had improvement in a different surrogate in a confirmatory trial, which led me to a less vigorous, but still painful facepalm. Apparently it did the same to the authors, who wrote in the discussion:
Studying the same surrogate efficacy measure that had been used to earn accelerated approval was considered sufficient by the FDA to confirm approval in certain cases, although it is not clear that such follow-up studies should be used as verification of benefit. Rather, a postapproval trial that uses the same surrogate measure as its primary end point should be described as corroborating the effect on the surrogate measure, perhaps in a larger or different patient population, unless the surrogate measure has been well validated. In other cancer drug approvals that we reviewed, the confirmatory trials used a different surrogate end point than the one used in the preapproval trial. In this situation, patients and physicians continue to lack information about whether the cancer drug improves survival or quality of life, which is essential in the benefit-risk evaluation for clinical decision making, unless the new surrogate is a validated surrogate.
Indeed. What was the point of using surrogate endpoints in a second trial, meant to be confirmatory? That doesn’t “confirm” anything either. As for the rest, five drug accelerated approvals (5%) have been withdrawn and an additional three (3%) did not demonstrate improvement in the primary end point in confirmatory trials. Five (5%) trials were delayed, 9 (10%) remain ongoing, 10 (11%) remain pending, and one each were terminated and released. Thus, a significant percentage of confirmatory studies are delayed or pending, which means that considerable time can elapse between the approval of a drug and the completion of its confirmatory trials.
The authors also note:
In describing the accelerated approval pathway, current FDA rules state that confirmatory trials of a drug should “verify and describe its clinical benefit, where there is uncertainty as to the relation of the surrogate end point to clinical benefit, or of the observed clinical benefit to ultimate outcome…such studies must also be adequate and well-controlled.” Although this language does not explicitly require that a confirmatory trial evaluate a clinical end point like OS, these rules do highlight that postapproval studies should be designed to resolve the uncertainty of the association between the surrogate measure and clinical benefit. This standard will be difficult to achieve via postapproval studies that use the same surrogate measures as those used in preapproval studies.
That last sentence is about as sarcastic a remark as you will ever find in a peer-reviewed medical study. Nice shade there, Dr. Gyawali!
In fairness, the authors do recognize the debate in oncology whether OS should remain the “gold standard” for oncology drug approval, or “whether achieving durable responses in single-arm trials should be sufficient to judge the clinical efficacy of a cancer drug”. The problem with this approach, from my perspective, is that many of these drugs are toxic, which means that a durable response could be at the cost of toxicity that negates (or worse, more than negates) any potential survival benefit attributable to the durable response.
There’s also a second study in the same issue of JAMA Internal Medicine by Chen et al, that looked at cancer drugs approved using only RR as the surrogate endpoint. (Shockingly, one-third of cancer drugs in the US are approved based only on RR.) Basically, they performed a comprehensive review of available package inserts for all oncology drugs that were FDA-approved on the basis of RR end points for any adult malignant disease from January 1, 2006, to September 30, 2018, including both accelerated and regular approvals. Of the 85 approvals that they examined, they reported that 14 (16%) had an RR less than 20%, 28 (33%) had an RR less than 30%, and 40 (47%) had an RR less than 40%, noting that “many cancer drugs are approved on the basis of low or modest RRs, typically in single-arm studies.” They noted in their conclusion:
Finally, many of these drugs have remained on the market for years without subsequent confirmatory data. When accelerated approvals based on RR were converted to full approval, 23 of 29 were made on the basis of surrogate end points (PFS or RR), 7 of 29 were made on the basis of RR, and only 6 of 29 were made on the basis of OS, an end point of clinical benefit. In summary, our analysis of drugs approved on the basis of RR end points suggests marked flexibility on behalf of the FDA to use this surrogate end point in the absence of randomized clinical trials.
These are shockingly low numbers.
What can be done?
An accompanying commentary by Richard Lehmann and Cary Gross notes that this is time of “unprecedented hope in the development of treatments for cancer.” noting that in comparison to many other regulatory agencies, the FDA “works to high standards of rigor and transparency” but that it “also works amid constant political clamor for faster access to innovative treatments.” They also note that these two studies “serve as a reminder that the accelerated approval pathway is a permissive process that tolerates nonrandomized trial methods and a variety of outcome measures that bear an uncertain relationship to patient benefit.” This is all true, unfortunately, and, as Chen et al show, there are a lot of drugs out there that undergo regular FDA approval based on surrogate endpoints and whose benefits in terms of OS are unproven. As Lehmann and Gross observe:
Such findings build on a growing body of work, which in aggregate, demonstrate a postmarketing evaluation process that is serving neither patients nor society well. First, the surrogates used for accelerated approval are poor predictors of clinically meaningful outcome. Second, study designs of postmarketing studies may not provide adequate information about comparative efficacy because some studies may not use appropriate control groups or rigorously account for treatment assignment bias. Third, there are concerns about whether postmarketing studies are being completed; it has been consistently reported that nearly half of postmarketing studies are delayed, and the proportion of completed studies remains low.1 Finally, nearly a quarter of these studies are not being publicly disseminated, even when completed.2
In other words, the postmarketing evaluation process is broken. As is noted in another accompanying editorial by Sarah DiMagno, Aaron Glickman, and Ezekiel Emanuel:
Second, using surrogate end points such as response rate to get drugs to patients quickly is only defensible if the studies’ findings are quickly evaluated in confirmatory postapproval trials that validly quantify overall risks and benefits, which inform physician and patient decision making. Manufacturers are required to conduct postapproval trials to ensure the signal from surrogate end points is real rather than noise.
Clearly, this is not being done nearly as fast or well as it should be. It’s a problem that needs to be fixed.
A complementary way to approach the problem is to find better surrogate endpoints and to work on assessing the correlation between the ones we use and clinical benefit in OS. As Gyawali et al note, their results, that only 20% of drugs approved based on surrogate endpoints improve OS, are consistent with other findings, and the use of these endpoints is not particularly rigorous. For example, a recent study by Kim and Prasad that found that in 56% of accelerated approvals and 37% of traditional approvals there was no formal analysis of the strength of the surrogate-survival correlation and that for accelerated approvals just four approvals (16%) were “made where a level 1 analysis (the most robust way to validate a surrogate) had been performed, with all 4 studies reporting low correlation (r≤0.7).” The conclusion? The use of surrogate end points for drug approval often lacks formal empirical verification of the strength of the surrogate-survival association. In another paper, Robert Kemp and Vinay Prasad noted:
We suggest this reliance on surrogates, and the imprecision surrounding their acceptable use, means that numerous drugs are now approved based on small yet statistically significant increases in surrogates of questionable reliability. In turn, this means the benefits of many approved drugs are uncertain. This is an unacceptable situation for patients and professionals, as prior experience has shown that such uncertainty can be associated with significant harm.
In other words, as I have argued before, if anything the FDA’s approval standards are arguably not rigorous enough, contrary to the arguments of anti-regulation ideologues who like to make ridiculous claims that the FDA is “killing” thousands of people by being too slow to approve life-saving new drugs or that you can replace the FDA approval process with, in essence, a “Yelp for drugs.”
I think DeMangno et al come closest to getting it right in their editorial when it comes to proposed solutions. Three problems are noted: that response rates are not meaningful clinical outcomes; that using surrogate endpoints to approve drugs is defensible only if the followup studies are done quickly and acted upon, which is not the case; and that the FDA still approves drugs even when the confirmatory data show no substantive benefit on clinically meaningful end points. They also note that there is no reason why the FDA has to rely on so many single trials using surrogate endpoints given that randomized trials in patients with metastatic disease could be done almost as quickly, given the shorter expected survival times. They then conclude:
Together, these 2 articles suggest that the ship of regulation has veered too far to one side, requiring at least 3 policy changes. First, the end point for confirmatory trials should never be the same surrogate end point used in the original study, and a new surrogate end point should be used only if there is a proven correlation between that end point and overall survival or improved quality of life. Most confirmatory trials should use overall survival and/or quality-of-life end points. Second, approval of drugs should be rapidly withdrawn when confirmatory trials report serious toxic effects or do not report meaningful clinical improvements. Finally, the confirmatory trials must be conducted promptly, with credible threats of reversed approval. Having more than a quarter of trials incomplete years after accelerated approval is unacceptable.
Lehmann and Gross note that postmarketing clinical trials are “expensive, requiring substantive infrastructure and oversight.” Of course, one argument for accelerated approval is that regular drug approval is too expensive and takes too long, implying that accelerated approval “gets cures to the patients” faster and for less money. Unfortunately, that observation can be countered by simply pointing out that many of these new drugs are nonetheless very expensive, and, as we have seen, too many of them are of marginal benefit at best, meaning that in far too many cases we pay a lot for not very much benefit. Clearly the FDA needs to do, at minimum, what DeMango et al suggest. Unfortunately, in the present political environment, all the political headwinds are pushing in a direction opposite of more rigorous standards for drug approval.