
{kind=link}
Last week, Steve Novella discussed announcements of drugs that, unlike the “miracle cures” (primarily hydroxychloroquine in 2020 and ivermectin this year) that have been promoted so heavily on social media, actually have evidence for their efficacy against COVID-19. Specifically, he mentioned Pfizer’s new drug Paxlovid, which in its clinical trials was 89% effective in preventing hospitalization due to COVID-19 if taken soon after symptoms develop. The other drug was Merck’s molnupiravir (trade name to be Lagevrio), which is now available through a clinical trial in the UK. Both drugs are very promising, and the Pfizer drug (a combination of PF-07321332 and ritonavir—more on why it’s a combination later—before it received a trade name), for example, decreased the hospitalization rate from 7% to 0.8% over 28 days in a Phase 2/3 study, which Steve correctly described as a “good absolute and relative risk reduction”, with no deaths in the drug treatment arm. (It was so good that Pfizer stopped the study, because it would be unethical to continue with such a result in an interim analysis.) As a result, Pfizer has applied for an emergency use authorization for its drug. In an interim analysis of a phase 3 study, the Merck drug demonstrated a 50% reduction in hospitalization, again with no deaths in the treatment arm.
Unsurprisingly, COVID-19 conspiracy theorists and antivaxxers are not pleased. This is why they seem to have come up with one of the sillier claims that I’ve seen thus far, although I must admit that it’s not obvious how silly they are if you don’t have a background in drug development and some specific knowledge about ivermectin and the Pfizer drug. (That’s rather the point.) Here are a couple of representative Tweets. I could post a whole bunch of them, but will restrain myself:
Protease inhibitors are established tools for preventing viral infections. The latest one from Pfizer inhibits an important protease used by the virus. One of the unmentionable compounds Ixx does that & more. Many other OTC compounds also do this. Why is this suppressed?
— pmcd (@pmcdunnough) November 12, 2021
And, of course:
John Campbell PhD explaining the similarities & differences between Ivermectin & Paxlovid.
Study of Uttar Pradesh (and how the media avoid India now that they're no longer a C19-disaster zone!) tells it all..
Meanwhile 100% of IC patients in GZA-hospitals in Antwerp are vaxxed. pic.twitter.com/WS0alaynDO
— miles. (@miles22402745) November 10, 2021
The basic idea behind this conspiracy theory is simple. Both ivermectin and the Pfizer drug are protease inhibitors, but ivermectin is being “suppressed” because Pfizer wants to make huge profits from its drug Paxlovid and ivermectin is a cheap competitor. (Merck’s drug is not a protease inhibitor, but instead works by being incorporated into the new RNA strand being copied to be packaged into the new virus particles being produced, where it, as one scientist put it, causes the coronavirus to “mutate itself to death.”)
As is the case with many COVID-19 conspiracy theories, there is a tiny grain of truth in the comparison of the drugs, but only a very tiny grain that conspiracy theorists seem to view as a whole field of wheat. In this post, I’ll discuss how one has to ignore some very basic pharmacological principles that differentiate the drugs in order to make the leap to the claim that ivermectin is just like Paxlovid, but being “suppressed” because it’s cheap and generic, as opposed to Paxlovid, which will be on patent and, of course, not cheap. In fact, ignoring one principle will do, the same pharmacological principle whose willful misunderstanding led to people claiming that ivermectin is a miracle cure for COVID-19, namely that concentration matters.
Spoiler alert: Ivermectin does inhibit the same protease that PF-07321332 does, but, as is the case for viral replication, it requires a concentration that is not achievable by oral dosing.
How does Paxlovid work?
As the conspiracy theorists point out correctly, Paxlovid belongs to a general class of drugs known as protease inhibitors. Enzymes belonging to the class of proteases break down proteins by specifically cleaving them into smaller peptides, often by recognizing a specific short amino acid sequence. (Indeed, back in my lab days, we used to use a trypsin to digest the proteins that held cells in cell culture to the substrate, so that we could replate them. Trypsin digests only at a very specific amino acid sequence.) There are a wide variety of proteases in nature, including serine proteases, cysteine proteases, metalloproteases, and more, with some working better in acid pH conditions, others in neutral or basic pH. These proteases are ubiquitous, as well, occurring in basically all organisms ranging from procaryotes to humans and are involved in a wide variety of physiological processes. The aforementioned trypsin, for instance, is a digestive enzyme normally found in the proximal small intestine.
It is therefore unsurprising that many viruses use proteases in their life cycles. The most famous of these among the general public is likely the human immunodeficiency virus (HIV), which causes acquired immunodeficiency syndrome (AIDS). It is the HIV protease that is the key target of the protease inhibitor cocktails that revolutionized the treatment of AIDS in the 1990s, changing it from a virtual death sentence to a chronic, manageable disease:
The main purpose of HIV is to copy itself as many times as it can. However, HIV lacks the machinery it needs to reproduce itself. Instead, it injects its genetic material into immune cells in the body called CD4 cells. It then uses these cells as a kind of HIV virus factory.
Protease is an enzyme in the body that’s important for HIV replication. Protease inhibitor drugs block the action of protease enzymes. This prevents protease enzymes from doing their part in allowing HIV to multiply, interrupting the HIV life cycle as a result. This can stop the virus from multiplying.
There are, of course, now a number of protease inhibitors approved for the use in treating HIV/AIDS, such as atazanavir, fosamprenavir, ritonavir, and others. In any event, protease inhibitors are the mainstays for the HIV/AIDS treatment known as HAART (highly active antiretroviral therapy), which usually consists of three or more medications to treat HIV, mixing protease inhibitors with other antiretroviral drugs. (Yes, some COVID-19 conspiracy theorists are noting that protease inhibitors are used to treat AIDS as well, and then circling back to an older—several months!—conspiracy theory that COVID-19 vaccines make you more susceptible to AIDS. (They don’t.)
Which brings us to PF-07321332.
PF-07321332 is a class of protease inhibitor that targets the 3C-like protease, which is the main protease found in coronaviruses. (Other names for this protease include coronavirus 3C-like protease, Mpro, SARS 3C-like protease, SARS coronavirus 3CL protease, SARS coronavirus main peptidase, SARS coronavirus main protease, SARS-CoV 3CLpro enzyme, SARS-CoV main protease, SARS-CoV Mpro and severe acute respiratory syndrome coronavirus main protease.) This particular protease has long been recognized as a potential drug target for the treatment of coronavirus-induced diseases because it plays a key role in processing the polyproteins translated from the viral RNA into functional viral proteins.
An article from Science explains in lay terms:
Monoclonal antibodies blocking surface proteins of the coronavirus can slow the infection of new cells with SARS-CoV-2, but their application has been hamstrung by cost, availability, and the need to infuse or inject them. In contrast, Pfizer’s ingestible candidate operates inside an infected cell; it blocks enzymes, called proteases, that normally act early in a virus’ life cycle to help it replicate (see graphic, below). Many protease inhibitors are approved for HIV treatment, and Pfizer’s compound has a nearly 20-year history. Pfizer scientists designed a version of the compound back in 2003 to block a protease in the coronavirus that causes severe acute respiratory syndrome (SARS), a cousin of SARS-CoV-2.
And:
As SARS-CoV-2 infects cells, reproduces itself, and spreads, the coronavirus relies on dozens of viral and host proteins to complete its life cycle. Pfizer’s new oral pill inhibits the main viral protease used to create other proteins for the virus. And Merck’s drug inserts a defective RNA building block when the virus uses an enzyme known as a polymerase to copy its genome.
And here’s a visual representation:
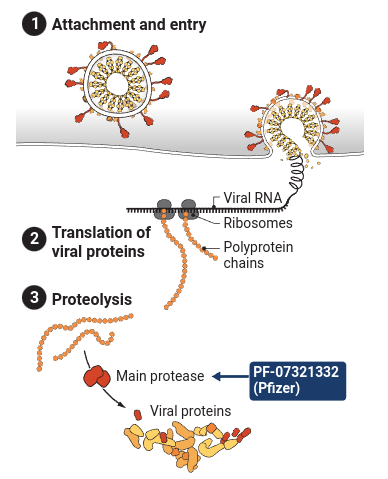
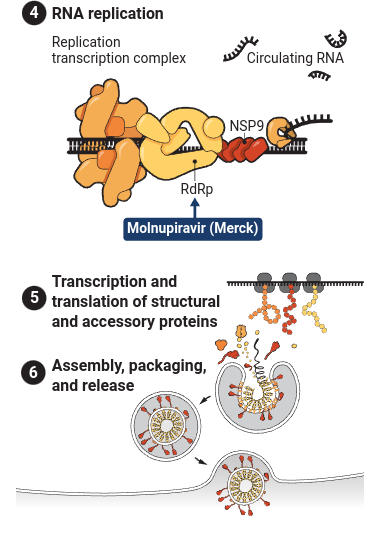
And another figure demonstrating how a long SARS-CoV-2 protein is cleaved into smaller structural proteins:
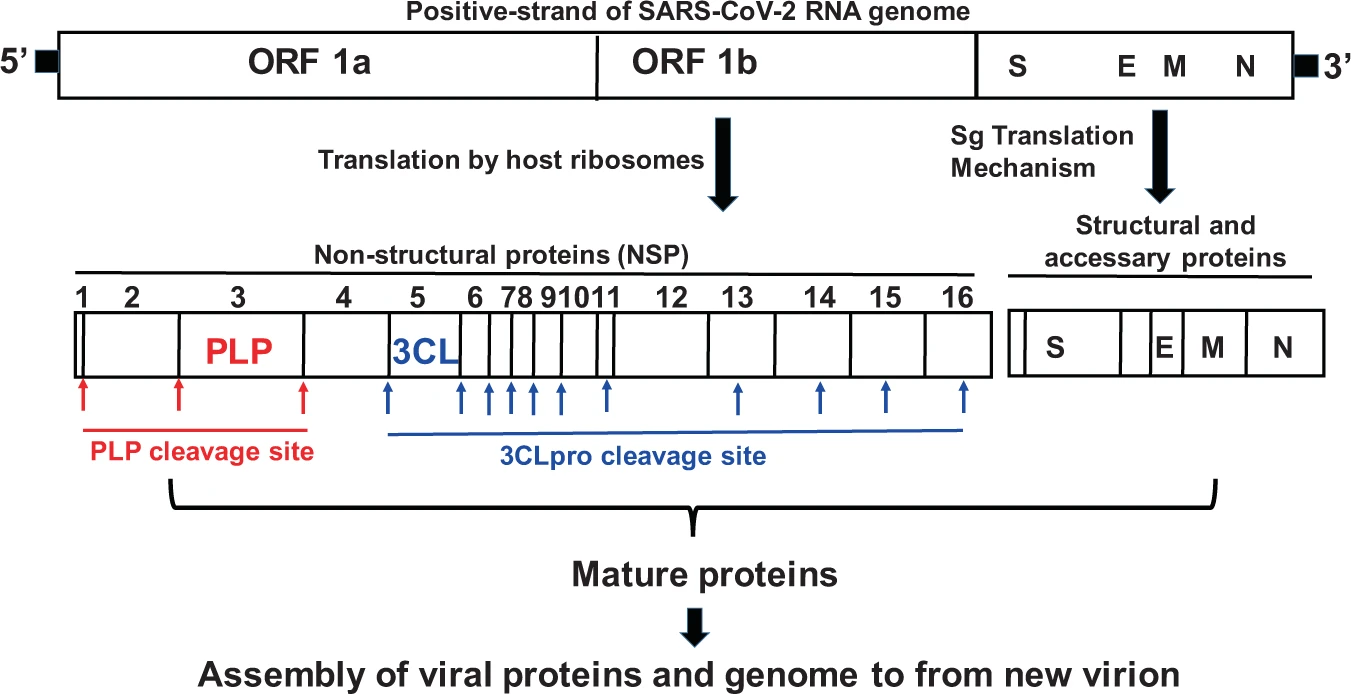
As Steve discussed last week, the rapid development of these two drugs adds more hope that the pandemic can finally be brought under control and become manageable, given that we have safe and effective vaccines and now drugs that can be used to treat the unvaccinated who become ill or the unfortunate vaccinated people who suffer breakthrough infections and, if real world data resemble clinical trial data, prevent the vast majority of them from dying or even requiring hospitalization.
It also turns out that PF-07321332 has been a long time coming. In the paper reporting the drug as a promising candidate to treat COVID-19:
In effort to identify inhibitors of the SARS-CoV-1 Mpro in response to the 2002 SARS outbreak led to the identification of PF-00835231 (1, Fig. 1) as a potent inhibitor of recombinant SARS-CoV-1 Mpro in a fluorescence resonance energy transfer (FRET)-based substrate cleavage assay (17). PF-00835231 also demonstrated potent inhibition (inhibition constant (Ki) = 0.271 nM) of recombinant SARS-CoV-2 Mpro, which is expected given that the SARS-CoV-1 and -CoV-2 Mpro share 100% sequence homology across their respective substrate binding sites (18).
Unfortunately, potent in vitro activity against a drug’s target does not necessarily translate to a good drug, and PF-00835231 had poor bioavailability. Specifically, it was poorly absorbed when given orally to experimental animals. So, as pharmacologists do when they have a potent drug candidate that isn’t well absorbed, Pfizer scientists started chemically modifying it to improve its oral bioavailability and testing those analogues in vitro for activity against the 3CL, and:
PF-07321332 demonstrated potent inhibition in FRET Mpro assays representing Mpro from all coronavirus types known to infect humans (6, 7, 30), including beta-coronaviruses (SARS-CoV-2, SARS-CoV-1, HKU1, OC43, MERS) as well as alpha-coronaviruses (229E, NL63) (Fig. 3B and table S2). No inhibitory effects were noted against several mammalian cysteine (caspase 2, cathepsin B, cathepsin L), serine (chymotrypsin, elastase, thrombin) and aspartyl (cathepsin D) proteases at the highest concentration tested (100 μM) of PF-07321332 (table S3). This was also the case for HIV-1 protease, a viral aspartyl protease (table S3).
That last part is important, because specificity is important. PF-07321332 would not have been very useful if, in addition to the SARS-CoV-2 protease, it also inhibited human proteases important in a number of processes, such as protein digestion. Such inhibition could have caused a number of undesirable and potentially harmful gastrointestinal side effects. PF-07321332 did not and was therefore deemed a good candidate with good oral bioavailability. It also exhibited potent antiviral activity against SARS-CoV-2 in vitro.
There was still a problem, though. There’s a reason that Paxlovid is a combination drug, containing PF-07321332 with ritonavir, an anti-HIV drug:
Paxlovid is a combination of two different drugs – the HIV drug ritonavir (a capsule) and an experimental drug PF-07321332 (a pill).
Ritonavir protects the body from metabolising PF-07321332. It acts by being broken down by the body first (known as a sacrificial chemical) to ensure enough PF-07321332 reaches the virus intact.
It’s a clever strategy to maximize the efficacy of PF-07321332. CYP3A4 is a P450 enzyme and often considered the most important drug-metabolizing enzyme, among the most abundant of its type in the liver. As noted by Pfizer:
These in vitro studies, which established a predominant role for CYP3A4 in the metabolism of PF-07321332, also presented an opportunity to boost therapeutic concentrations of PF-07321332 in the clinic via co-dosing with the potent CYP3A4 inactivator ritonavir (RTV), which is used as a pharmacokinetic enhancer of several marketed protease inhibitors (e.g., darunavir, lopinavir) that are subject to metabolic clearance via CYP3A4 (35, 36).
Before I move on, I will note that drug companies like Pfizer often have huge libraries of compounds predicted to target certain enzymes based on computer modeling, and we can expect more compounds like this in the future to be tested.
Speaking of computer libraries, let’s contrast Pfizer’s Paxlovid with ivermectin.
“Ivermectin works just like the Pfizer drug!” Not so much.
So where does the claim that Paxlovid is just “repackaged ivermectin” designed to increase pharma profits come from? Again, it reminds me a lot of how ivermectin came to be thought of as a “miracle cure” for COVID-19 based mainly on in vitro studies of its ability to inhibit SARS-CoV-2 replication cell culture. To that end, let’s look at the paper from which I took one of the figures above. Basically, it was a computer modeling study that was used to identify existing compounds that could target 3CLPro, hence the segue I used. The authors used computer modeling to identify potential promising candidates to inhibit 3CLPro and then tested the top “hits” in cell culture. One of the compounds identified by this strategy was ivermectin.
Before I get to this, let me just point out that it was thought that ivermectin inhibited SARS-CoV-2 replication through the inhibition of a different protein, α/β1 importin, something reported a decade ago. This particular protein is involved in the transport of proteins into the nucleus from the cytoplasm. In the case of SARs-CoV-2 infection, the transport of certain proteins into the nucleus is important for completion of its lifecycle. (I won’t go into the details other than that.)
Now here’s the thing. Inhibition of this protein complex by ivermectin and preventing the replication of HIV and the Dengue virus requires fairly high concentrations (at least in terms of drug concentrations). The original Australian paper that examined the ability of ivermectin to inhibit SARS-CoV-2 replication showed similar results. Now the problem. As stated in this review:
As noted, the activity of ivermectin in cell culture has not reproduced in mouse infection models against many of the viruses and has not been clinically proven either, in spite of ivermectin being available globally. This is likely related to the pharmacokinetics and therapeutic safety window for ivermectin. The blood levels of ivermectin at safe therapeutic doses are in the 20–80 ng/ml range [44], while the activity against SARS-CoV2 in cell culture is in the microgram range. Ivermectin is administered orally or topically. If safe formulations or analogs can be derived that can be administered to achieve therapeutic concentrations, ivermectin could be useful as a broad-spectrum antiviral agent.
Even this paper proposing ivermectin as a treatment for COVID-19 notes:
A dose of 12 mg twice daily alone or in combination with other therapy for 5–7 days has been proposed as a safe therapeutic option for mild, moderate or severe cases of Covid-19 infection.10 The time to reach maximum plasma concentration of 20–50 ng/ml, after a dose of 6 or 12 mg, respectively is approximately 4 h.
This was almost certainly the reason that ivermectin doesn’t work against COVID-19 in spite of its activity in vitro against SARS-CoV-2. It requires a concentration 10- to 20-fold higher than is safely achievable in the blood. That’s why the review concluded that maybe an ivermectin analogue that is either more active or can achieve a higher concentration safely in the bloodstream is worth investigating. Ivermectin itself is a crap drug based on that mechanism.
But what about its protease inhibition? It’s the same damned principle. Yes, ivermectin can inhibit 3CLPro in vitro, but it’s the same old story, as has been pointed out:
There is a tiny iota of truth: There *is* evidence that both drugs bind/inhibit the viral main protease (Mpro).
But there is a critical difference between them:
For PF-07321332 this is at a concentration actually achievable in vivo (3nM), ivermectin isn’t even close (21μM)!
2/ pic.twitter.com/m21jkKZDv1— Nick Mark MD (@nickmmark) November 8, 2021
The papers to which Nick is referring are the Pfizer paper, recently published and no longer a preprint, and the computer modeling paper.
I further note that these efforts to target 3CLPro date back to 2003 during the original SARS epidemic:
I am told (in general) that they have large libraries of enzymes / protease inhibitors they scan through (in silicon). They then try to find a pre-existing one with properties similar to what they want, then start modifications and testing from there to get a better match.
— Punctuated Equilibrium 🦠x0 (@GainOfDystopia) November 6, 2021
To put things in full context, here’s a reminder. Nanomolar (nM, 10-9 mol/L) concentrations are 1,000-fold lower than micromolar (μM, 10-6 mol/L). The idea is the same for 3CLPro as it was for α/β1 importin. The concentrations of ivermectin needed to inhibit 3CLPro are much, much higher than can be safely achieved in the blood and extracellular fluids of human beings with oral dosing, and it that’s what needs to happen for the drug to work in a living, breathing human being. Many are the drugs that show good activity in cell culture but founder when the attempt is made to test them in mice and other laboratory animals. It turns out that this is just one (among many) reasons why drug development is very, very hard and that promising compounds that appear to work well in cell culture fail when tested in animals.
Indeed, let’s just say that a Ki (the concentration producing 50% of maximal inhibition of a target) of 3.1 nM for a target like 3CLPro is the sort of incredibly low Ki that pharmacologists and medicinal chemists dream about when trying to develop a new drug to target a specific enzyme. I myself have fallen victim to the very same problem in my research career so I am very, very aware of this problem. Let’s just say that my drug’s Ki was a lot closer to that of ivermectin than that of PF-07321332. Let’s also say that my drug, like ivermectin, shows a lot of other fairly nonspecific activities that require high concentrations as well and leave it at that. Again, drug development is hard, and promising compounds that work in cell culture often fail for reasons of achievable plasma concentrations and pharmacokinetics, something that anyone who’s done drug development is painfully aware of.
Unfortunately, apparently ivermectin cultists are not aware of this very basic principle, which is why the drug was promoted based on an Australian cell culture study that in and of itself strongly suggested that the drug was actually not a particular promising COVID-19 treatment based on pharmacokinetic considerations alone with respect to its ability to inhibit viral replication and α/β1 importin. Now they’re doing it again based on a study that shows that ivermectin is not a particularly promising COVID-19 treatment based on pharmacokinetic considerations alone, just this time with a different target, 3CLPro.
Contrary to the conspiracy theory being pushed by cranks, Paxlovid is not just “repackaged” ivermectin, and ivermectin isn’t being “suppressed” to protect the profits of Pfizer. No one, least of all I, denies that large pharmaceutical companies have done some pretty shady things, but “suppressing” ivermectin in favor of the Pfizer and Merck drugs does not appear to be one of them. Ivermectin, although a fantastic drug to treat diseases caused by roundworm infestations, just doesn’t work against COVID-19, and there was never any really compelling reason to suspect that it would. Now here’s hoping that the clinical trial results for PF-07321332 (Paxlovid) and molnupiravir (Lagevrio) hold up in the real world. That would truly be as much of a game-changer as the development of effective and safe vaccines a year ago.