{kind=link}
Author’s note: I have received messages informing me about errors that were made in haste during the writing of this article. I have corrected them and the specific changes made can be found in the Erratum. In addition, I have also received quite a few questions asking where more can be learned about MPNs. I have included a short list of resources for both the lay-person and the professional. I thank all readers for pointing out true factual error. It helps all of us.
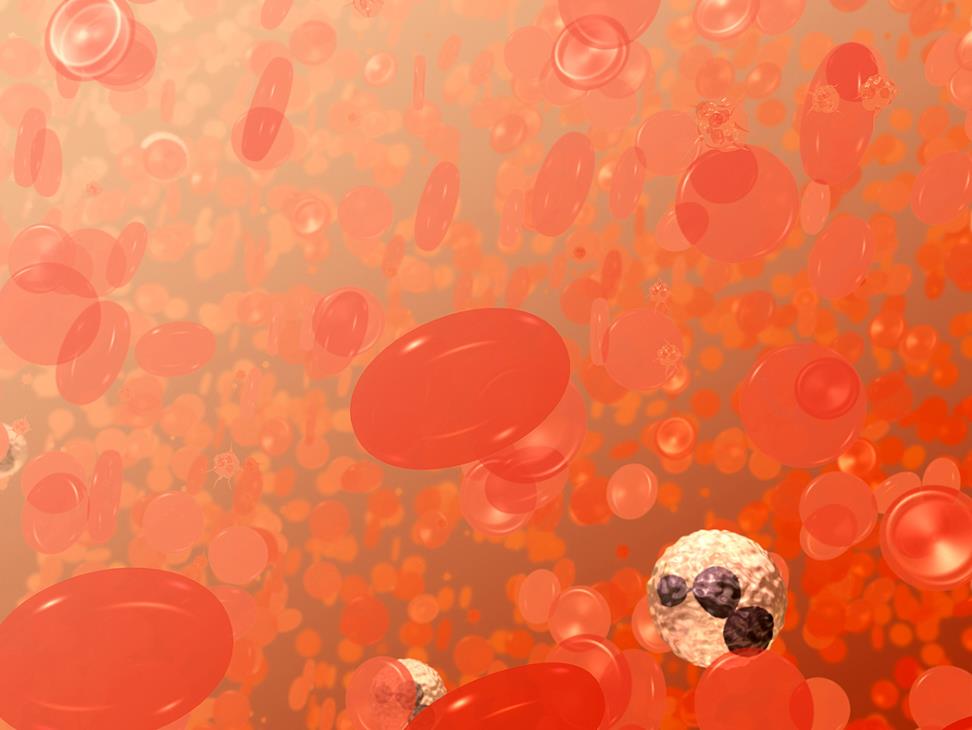
A field of blood cells. The bi-concave disks are red blood cells or erythrocytes. The white cell with the dark purplish, multi-lobed nucleus is a neutrophil, a type of white blood cell or leukocyte. The smaller spikey objects are platelets. Image and text adapted from the NCI Visuals Online Library image 3696, created by Donald Bliss.
Most people will be familiar with the more common blood cancers like leukemias and lymphomas, but there is a rarer group that still causes considerable suffering. Cancers in this group are in many ways harder to treat, and due to their rarity they receive little research funding and even less recognition. These cancers are called myeloproliferative neoplasms (MPNs). This post will deal with what are called “classical” (or “typical”) Ph-negative MPNs. Ph stands for the Philadelphia chromosome, a translocation that is the defining feature of chronic myeloid leukemia (CML). Its absence is part of the diagnostic criteria for the Ph negative MPNs. I have a vested interest in these cancers as I have a rare MPN myself, which I will discuss later.
Basic hematology and MPN fundamentals
All blood cells form in the bone marrow from pluripotent stem cells and then further differentiate into two types of blasts; myeloid blasts and lymphoid blasts. Myeloid blasts become red cells, platelets, and all white cells except lymphocytes. Lymphoid blasts become lymphocytes, B-cells, T-cells, and plasma B cells.
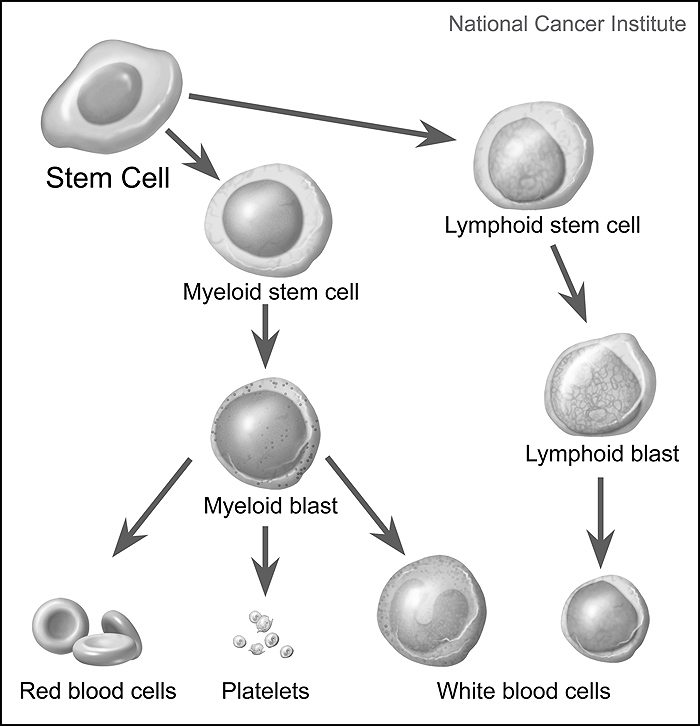
Simplified diagram of stem cells maturing into one of three types of mature blood cells: red blood cells, platelets, and white blood cells. Precursor cells are also shown: stem cells, myeloid blasts, lymphoid stem cells, and lymphoid blasts. Image and text adapted from the NCI Visuals Online Library item 4348, created by Alan Hoofring.
MPNs originate in the myeloid line from various mutations and affect the bloodlines in varying ways. It is best to consider these cancers as occupying a spectrum rather than being discrete entities. MPNs don’t have stages per se, but phases. They have the propensity to evolve into more severe classifications, then finally into acute myeloid leukemia (AML), a form that is notorious for its resistance to treatment. This is sometimes called “MPN Blast Phase”. The incidence of transformation varies between MPNs as well as the average life expectancy of those diagnosed. The following are the “typical” MPNs and my own.
- Essential thrombocythemia (ET): The most indolent of MPNs, causes an increase in the platelet count that sometimes cross >1,000 x 109/L (the average is about 150-450 x 109/L). The main concern is thrombosis (and paradoxically, also bleeding at platelet levels above 1500 x 10^9/L). Life expectancy varies between studies, but is on par with to slightly below the average population. The prevalence is 38-57 cases per 100,000 persons.
- Polycythemia vera (PV): This primarily causes an increase in red blood cells but is often accompanied by increases in platelets and white cells. These significantly increase the risk of major bleeding and clotting problems. Median survival based a recent large-scale study is 11-28 years. The prevalence is 44-57 cases per 100,000 persons.
- Myelofibrosis (MF, including post-PV and post-ET as well): Causes the tissues of the bone marrow to scar and become incapable of producing new blood cells. This often results in cytopenias (low blood counts) that can lead to lethal infections and an enlargement of the spleen (sometimes requiring removal) and liver through a process known as “extramedullary hematopoiesis”, which means that the creation of blood switches partially to other organs, like the liver and spleen, to make up for the loss of marrow production. Median survival is roughly six years. The prevalence is 4-6 cases per 100,000 persons.
- Myeloproliferative neoplasm/Myelodysplastic syndrome, unclassified (MPN/MDS-U): Acts like both an MPN and MDS (where the bone marrow does not produce enough of some blood cell types). MPN/MDS-U may present in varying ways, showing features of just a few or many distinct classifications. (I show features of both PV and MDS-U.) It is essentially a catch-all when no more specific diagnosis can be provided. It is too rare for an incidence, prevalence, or prognosis to be estimated.
In addition to the risks of death from bleeding, thrombosis, infections, and evolution to other classifications and AML, all MPNs carry a high symptom burden, with many patients experiencing symptoms of severe fatigue, inactivity, night sweats, bone pain, and difficulty concentrating (among others). To make matters worse, treatment options are limited.
Current treatment options
In the world of MPNs there are few treatment options, remissions are extremely rare, and this group of cancers is often considered incurable. Depending on the type, there are several options for management; most have been around since the 1950s. The oldest of these is therapeutic phlebotomy for PV; this is medical lingo for actual bloodletting. The process is exactly the same as giving blood, with a goal of bringing the hematocrit level below 45% for men and 42% for women. The frequency of this action varies from patient to patient; some a few times a year, some a few times a month. Another mainstay is simple aspirin therapy to help prevent blood clots. If these first-line treatments fail (most often intolerance) or a patient is in a worse prognostic group (often due to a previous clot) then pharmaceutical cytoreductive therapy is initiated.
This is essentially chemotherapy, the most common being a drug called hydroxyurea/hydroxycarbamide. It is thought to act through RNA inhibition and reduces the blood counts. Busulfan or similar drugs (alkylating agents) may sometimes be used, but this is rare after it was discovered that it may actually speed up the evolution to AML and is typically only recommended in older patients. Other drugs occasionally used include anagrelide for platelet reduction and radioactive phosphorus (P32) which reduces all types of blood cells. None of these are ideal, however, as they have severe adverse effects including the possible speeding of progression.
As a last-ditch effort, sometimes an allogenic stem cell transplant (allo-SCT) is undertaken. It is often viewed as the only potentially curative option for MPNs. The risk of death is often uncomfortably high (> 50% in some cases); in addition, the patient’s age and other comorbidities may be contraindications. There is no guarantee of long-term survival, as MPN patients are at a greater risk for secondary malignancies and there is always a risk of relapse.
In the future
It is, however, not all doom and gloom for MPN patients. Even with limited research funding, there is a small but highly dedicated group of doctors and researchers that have been developing and testing new options. Most recently this has led to the approval of ruxolitinib (trade name Jakafi), the first drug approved for both MF and PV. It works through targeting the pathways affiliated with a mutation common in MPNs called JAK2 (V617F) by inhibiting JAK1 and JAK2 enzymes. It has been shown to be effective in reducing constitutional symptoms in patients with both diseases, as well as reducing the phlebotomy requirement for PV patients.
Another drug showing promise that is currently off-label but in late phase trials is peglyated interferon. It has the advantage of being a non-chemotherapeutic agent and works through modulating the immune system. It has even been shown to induce hematologic remission in some patients (though this is not the same as a cure). The drug is often the first choice for young, high-risk patients, but long-term adherence is sometimes hindered by adverse effects, including flu-like symptoms and suicidal ideations.
Newer drugs are following the path of targeted therapies such Jakafi. There are several other JAK inhibitors in trials at multiple centers worldwide. Another possible route is telomerase inhibitors, such as imetelstat. Telomerase in a chemical in the body that basically helps prevent premature degradation of our telomeres which cap the ends of chromosomes (and a frequent target of quackery). However, in cancer this is obviously not a good thing. And with the advent of CRISPR-CAS9 gene editing there seems to hope to go beyond merely controlling the disease and actually arriving at a cure, but I must say that such options are sadly many years away from being a realistic possibility.
My experience with myeloproliferative neoplasms
I was initially diagnosed with an MPN-U, later revised to an MPN/MDS-U in the autumn of 2014. It took approximately six months to reach this diagnosis from a visit to my GP for headache and fatigue. After my initial tests, I was met with innocuous diagnoses of a mild infection and a kidney cyst. Deeper testing revealed rarer results that led me to being referred to pretty much every type of clinical specialist out there, along with meeting a substantial population of New York City’s hematologists. I was screened and tested for various cancers including brain, lung, colon, stomach, thyroid, kidney, liver, pretty much all of them. It wasn’t until I landed at Memorial Sloan Kettering’s leukemia service that the idea of my actual diagnosis was entertained. It was during the 2nd or 3rd visit that it was confirmed and I was immediately started on cytoreductive therapy. Prior to that I was averaging 2-3 phlebotomies a week (that’s a liter and a half of blood a week).
The fun didn’t end there, it was discovered that I quickly became resistant to the chemo after just a few months. Over the past few years I have been on three different agents and I am now at the point where I have reached the maximum possible dose on the last line therapy. I have been seen at Dana-Farber, Fred Hutch, and had my case reviewed by every expert my doctor and I could think of. There aren’t any easy solutions.
I experience multiple symptoms daily, both from the cancer itself and my medications. I have severe fatigue, nausea, headaches, night sweats, muscle aches, and trouble concentrating. Not three weeks ago I had a transient ischemic attack (TIA or “mini stroke”). As I result I now take an increased dose of aspirin and a statin in addition to my 10 other medications. I don’t know if they will help me, but I feel it is the safest option.
Right now, we are looking for other medications that have cytoreductive effects with minimum side effects; however, the issue is that most chemotherapeutic drugs are not intended to be used constantly for years at a time. I have also been evaluated for an allo-SCT, but that carries all the risks I mentioned above, and most transplant hematologists are hesitant to do it, as am I.
I don’t ask readers for pity though: I ask that you keep MPNs in mind along with other rare cancers. It is not simply lung or breast cancer patients that need attention, but all those with cancer. It has been shown that roughly 20% of all cancers diagnosed are actually rare cancers. Even eight years after the reclassification of MPNs as a type of cancer, the American Cancer Society does not recognize them on their website or support services.
I also would like to say that if it wasn’t for the strict attention to science that hematology is intertwined with today, I wouldn’t be alive to write this today. Science-based medicine can and does save lives. I’m not sure what the cure for MPNs or me is, but I’m sure it’s not in a turmeric smoothie or the hands of a reiki master.
Patient resources
- http://www.mpnresearchfoundation.org/
- http://www.cancerresearchuk.org/about-cancer/other-conditions/myeloproliferative-neoplasms
- http://www.lls.org/myeloproliferative-neoplasms
- https://www.cancer.gov/types/myeloproliferative
A few technical resources I like
- Williams Hematology, 9E by Kenneth Kaushansky Dr. MD
- Wintrobe’s Clinical Hematology, Thirteenth Edition by John P. Greer MD
- Hematology: Basic Principles and Practice, 6th by Ronald Hoffman. (new edition comes out in a few days, I think it’s well worth it. Hoffman is a great hematologist and a great MPN expert)
- www.bloodjournal.org
- www.nejm.org
- http://www.haematologica.org/
- www.readbyqxmd.com (program that allows you link with all your journals or by keyword and automatically receive new articles to your devices with an option to download full pdf)
- https://www.uptodate.com/home
- http://www.dynamed.com/home/
Erratum
- Corrected an error in the listing of MPNs that had caused confusion and made unclear whether prevelance or incidence was being discussed.
- Corrected blood value measurements because of error in essential thrombocythemia.
- Modified ET to include paradoxical bleeding.
- Clarified the mechanism of action of Jakafi.