
{kind=link}
Why haven’t we cured cancer yet? How many times have I heard that question before, and how many times have I tried to answer it? I don’t know. I do know that I’ve pointed out many times that cancer is not one disease, but hundreds and that the genomes of cancers are messed up, real messed up, and that trying to cure any single cancer is fighting against the power of evolution that makes cancer more resistant to treatment the longer it’s exposed to a given treatment. Still, every so often a study comes out that leads me to revisit the question in light of the new data published. This time around, it’s a study published earlier this month in Nature by the Pan-Cancer Analysis of Whole Genomes Consortium (PCAWG) of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA) examining the evolutionary history of over 2,600 cancers representing 38 different kinds of cancer, each of whose whole genome was sequenced and analyzed. The conclusions of the study, the work of a more than a thousand scientists in 37 countries on four continents taking place over more than a decade, are fascinating and very much consistent with cancer being primarily a disease caused by mutations in a relatively small set of genes. Actually, it’s a series of papers based on the same massive study. Before we dig in, let’s consider just how massive this undertaking was:
The project involved an interdisciplinary group of scientists from 4 continents, with 744 affiliations between them, who had to overcome major technical, legal and ethical challenges to carry out distributed analyses while protecting patient data. Researchers were divided into 16 working groups, each focused on distinct facets of cancer genomics — assessing the recurrence of mutations, for instance, or inferring tumour evolution.
Altogether, the consortium performed integrative analyses of 38 tumour types. The group sequenced 2,658 whole-cancer genomes (Fig. 1), alongside matched samples of non-cancerous cells from the same individuals. These data were complemented by 1,188 transcriptomes — the sequences and abundances of RNA transcripts in a tumour.
These efforts involved extensive quality control and coordinated data processing, as well as massive, systematic experimental validation of the computational pipelines used to detect mutations. Many computational algorithms and pipelines were used and compared in concert. This required hundreds of terabytes of data, spread across multiple data centres, and probably millions of processing hours — all facilitated by cloud computing. Notably, the PCAWG efforts provide a prime example of how cloud computing can make international collaboration possible and help to advance data-intensive fields.
These sorts of projects just weren’t possible not too long ago. Neither the computational power nor the techniques needed to sequence an entire genome in a short period of time existed. The results of this project were published in six papers:
- Pan-cancer analysis of whole genomes
- Analyses of non-coding somatic drivers in 2,658 cancer whole genome
- The repertoire of mutational signatures in human cancer
- Patterns of somatic structural variation in human cancer genomes
- The evolutionary history of 2,658 cancers
- Genomic basis for RNA alterations in cancer
In addition to these papers, a number of companion papers were published in other Nature Publishing Group journals, so many that I must confess that I haven’t had a chance to read them all yet. That’s why I’ll mainly focus on the core six listed above, mainly (but not exclusively) the first and fifth papers, because the effect of evolution on cancer development has always been an interest of mine.
Personally, I like how the first paper mentions that “cancer” is not a single disease, that it’s an incredibly diverse collection of diseases:
‘Cancer’ is a catch-all term used to denote a set of diseases characterized by autonomous expansion and spread of a somatic clone. To achieve this behaviour, the cancer clone must co-opt multiple cellular pathways that enable it to disregard the normal constraints on cell growth, modify the local microenvironment to favour its own proliferation, invade through tissue barriers, spread to other organs and evade immune surveillance21. No single cellular program directs these behaviours. Rather, there is a large pool of potential pathogenic abnormalities from which individual cancers draw their own combinations: the commonalities of macroscopic features across tumours belie a vastly heterogeneous landscape of cellular abnormalities.
This heterogeneity arises from the stochastic nature of Darwinian evolution. There are three preconditions for Darwinian evolution: characteristics must vary within a population; this variation must be heritable from parent to offspring; and there must be competition for survival within the population. In the context of somatic cells, heritable variation arises from mutations acquired stochastically throughout life, notwithstanding additional contributions from germline and epigenetic variation. A subset of these mutations alter the cellular phenotype, and a small subset of those variants confer an advantage on clones during the competition to escape the tight physiological controls wired into somatic cells. Mutations that provide a selective advantage to the clone are termed driver mutations, as opposed to selectively neutral passenger mutations.
The fifth paper also points out how important evolution and selective pressures are on the development of any given cancer:
Similar to the evolution in species, the approximately 1014 cells in the human body are subject to the forces of mutation and selection1. This process of somatic evolution begins in the zygote and only comes to rest at death, as cells are constantly exposed to mutagenic stresses, introducing 1–10 mutations per cell division2. These mutagenic forces lead to a gradual accumulation of point mutations throughout life, observed in a range of healthy tissues5,6,7,8,9,10,11 and cancers12. Although these mutations are predominantly selectively neutral passenger mutations, some are proliferatively advantageous driver mutations13. The types of mutation in cancer genomes are well studied, but little is known about the times when these lesions arise during somatic evolution and where the boundary between normal evolution and cancer progression should be drawn.
I’ve discussed the heterogeneity of cancer before based on much smaller studies from several years ago. Basically, because of mutations and different selective pressures on a given cancer, the cells in one part of a cancer are often quite different than the cells in another part, and the cells in metastases are, again, often quite different from the cells in the primary tumor mass. One time when I described the results of a study sequencing cancer genomes, I described these genomes as looking like someone threw a miniature grenade into the nucleus of the cell from which the cancer originated. In other words, these are some really messed up genomes. (I wanted to use another word to describe it, but this is a family blog—sort of, anyway.) I used this example to explain once again that cancer is not a single disease. It’s hundreds of diseases. Although there are common themes in how cells become cancerous, such as loss of responsiveness to growth signals with a resultant ability to grow unchecked, evasion of programmed cell death (apoptosis), inducing the surrounding tissue to provide a blood supply (angiogenesis), evading the immune system, and invading the blood or lymphatic systems to travel elsewhere in the body and take up shop in other organs, such as liver, lung, or bone, individual cancers acquire these necessary (to the cancer) abilities through many different mechanisms. The current set of studies helps describe the sorts of genomic changes in cancer cells that allow them to do these things.
The first key finding, reported in the first paper, is that on average each cancer genome contains four or five driver mutations, which provide the cells with a selective advantage. The authors’ initial analysis found nearly 10% of cancers did not have an identifiable driver mutation. When they went back and accounted for technical issues (some normal matched samples had significant contamination with DNA from cancerous samples) and other factors that could hamper identification of driver mutations, they still were unable to identify a driver mutation in 5.3% of the cancer genomes studied, which tells us that we have not yet identified all the mutations that can drive cancer.
The authors also looked at the types of mutational events observed in the cancers. In particular they looked at the sorts of mutations that can generate multiple mutations in a single catastrophic event: (1) chromoplexy, in which repair of co-occurring double-stranded DNA breaks—usually on different chromosomes—results in shuffled chains of rearrangements; (2) kataegis, a focal hypermutation process that leads to locally clustered nucleotide substitutions, biased towards a single DNA strand; and (3) chromothripsis, in which tens to hundreds of DNA breaks occur simultaneously, clustered on one or a few chromosomes, with near-random stitching together of the resulting fragments. Chromoplexy and translocations were found in 18% of samples and were particularly prominent in prostate adenocarcinoma, lymphoid malignancies, and, unexpectedly, thyroid adenocarcinoma. Kataegis events were found in 60.5% of all cancers, with particularly high abundance in lung squamous cell carcinoma, bladder cancer, acral melanoma and sarcomas. Finally, chromothripsis was observed in 22.3% of the samples, most frequently among sarcoma, glioblastoma, lung squamous cell carcinoma, melanoma and breast adenocarcinoma.
At least as important as the types of mutations observed in cancer is the timing of such mutations, which is what the fifth paper is all about. It turns out that you can reconstruct part of the evolutionary history of a cancer through whole genome sequencing:
The genome of a cancer cell is shaped by the cumulative somatic aberrations that have arisen during its evolutionary past, and part of this history can be reconstructed from whole-genome sequencing data3 (Fig. 1a). Initially, each point mutation occurs on a single chromosome in a single cell, which gives rise to a lineage of cells bearing the same mutation. If that chromosomal locus is subsequently duplicated, any point mutation on this allele preceding the gain will subsequently be present on the two resulting allelic copies, unlike mutations succeeding the gain, or mutations on the other allele. As sequencing data enable the measurement of the number of allelic copies, one can define categories of early and late clonal variants, preceding or succeeding copy number gains, as well as unspecified clonal variants, which are common to all cancer cells, but cannot be timed further. Lastly, we identify subclonal mutations, which are present in only a subset of cells and have occurred after the most recent common ancestor (MRCA) of all cancer cells in the tumour sample (Supplementary Information).
The ratio of duplicated to non-duplicated mutations within a gained region can be used to estimate the time point when the gain happened during clonal evolution, referred to here as molecular time, which measures the time of occurrence relative to the total number of (clonal) mutations. For example, there would be few, if any, co-amplified early clonal mutations if the gain had occurred right after fertilization, whereas a gain that happened towards the end of clonal tumour evolution would contain many duplicated mutations14 (Fig. 1a, Methods).
The following figure from the paper gives you an idea of the principles involved, in particular panel A.
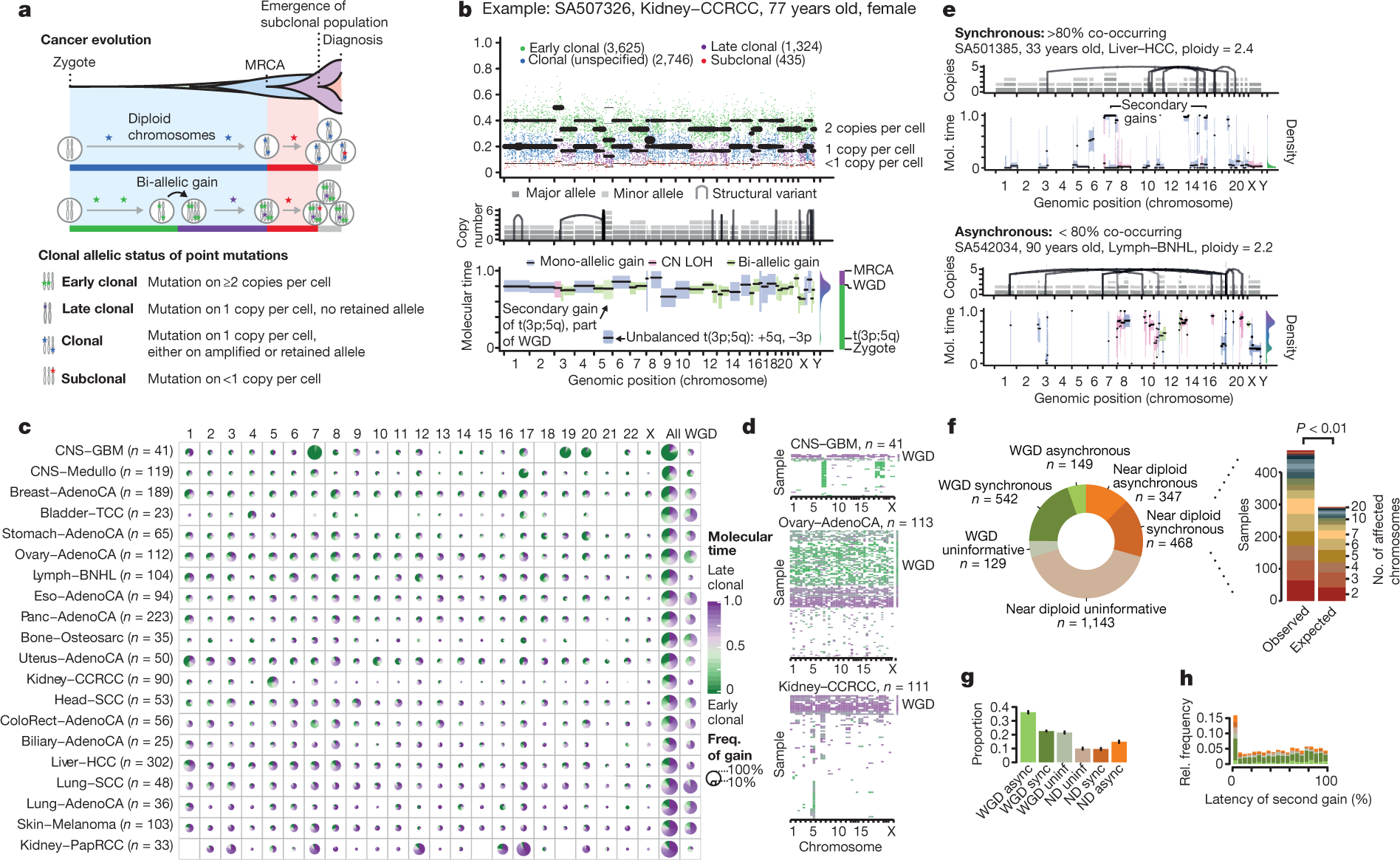
The results of this particular analysis shows that, unsurprisingly, driver mutations that most commonly occur in a given cancer also tend to occur the earliest in the development of that cancer. In a similar vein, if a certain cancer type is characterized by copy number gains, these copy number amplifications tend to occur early as well. One example discussed is clear cell cancer of the kidney, which frequently has a copy number gain of part of chromosome 5, which tends to arise early in the evolution of this particular cancer. In addition, the authors found that mutational signatures change over time in at least 40% of tumors and tend to reflect an increase in the frequency and severity of DNA repair defects.
Perhaps the most important finding of this study is its finding that driver mutations can occur years before cancer is diagnosed. Figure 6 (below) shows the order of mutations for four common cancers, colorectal cancer, lung cancer, ovarian cancer, and pancreatic cancer:
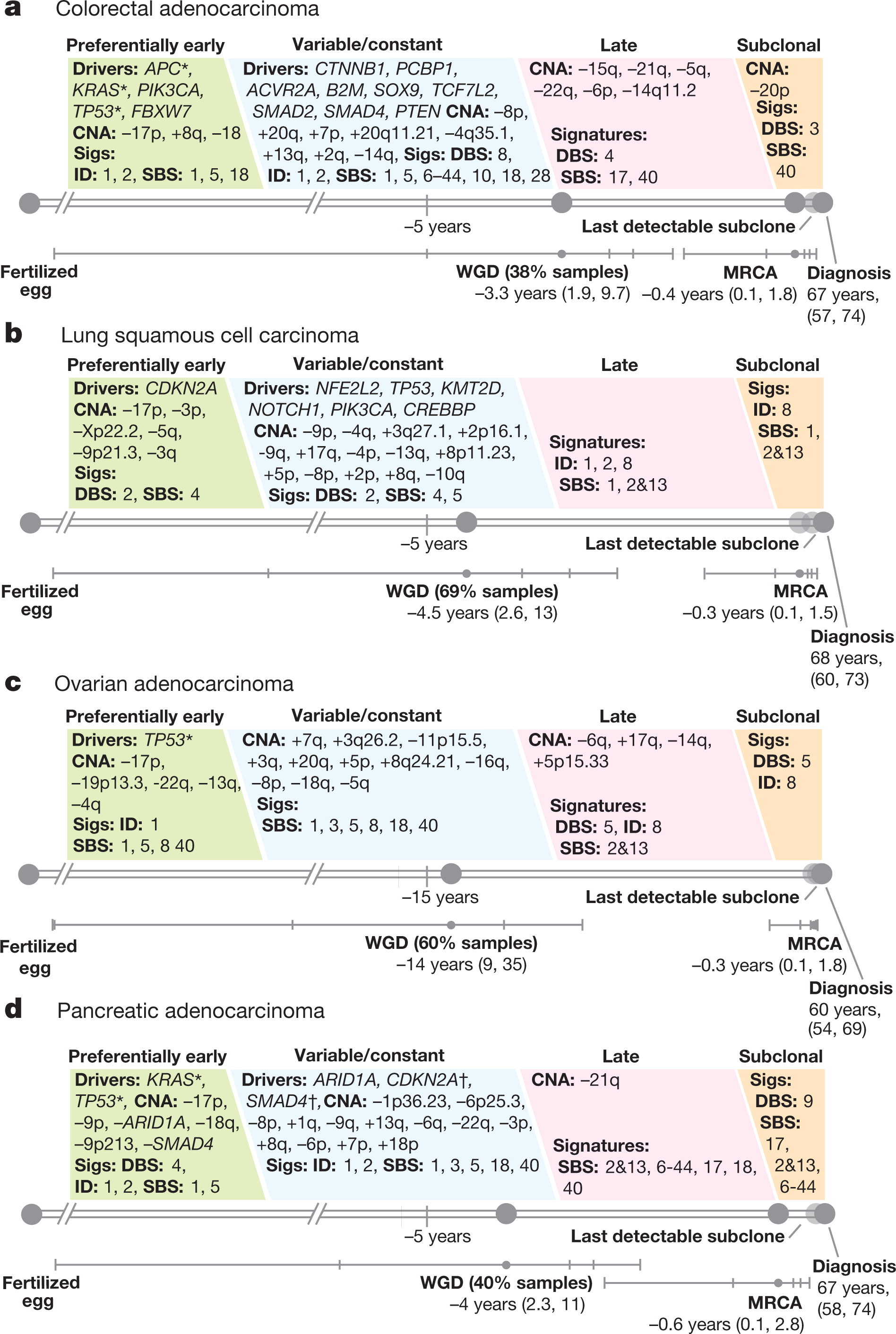
Another key finding of these analyses, reported in the second paper, is that it’s not just mutations on coding regions (i.e., regions of DNA that contain genes that produce proteins) that can drive cancer. Although a lot less common than driver mutations in coding regions, driver mutations in noncoding regions do occur. Usually, these are mutations in regulatory regions of the genome that result in the overexpression of a protein. This sort of analysis is much harder because it is a lot more difficult to accurately detect mutations in noncoding regions than in coding regions, or to assess their recurrence, which is one reason why, although it has been known that such mutations exist, their prevalence in cancer has not been well characterized. Great care had to be taken to exclude artefacts and find such mutations. In this study, the authors report finding relatively frequent mutations in non-coding regions of the telomerase gene TERT that result in overexpression of the telomerase enzyme (which can allow tumor cells to divide uncontrollably by eliminating the limit on the number of times a given cell can divide), that is similar to the high prevalence of telomerase mutations found in a previous pan-cancer study of more-advanced (metastatic) tumors, namely 12%. Although the study could not conclusively rule out the existence of other non-coding drivers, it did show that driver mutations in noncoding parts of the genome are not nearly as common as such mutations in coding regions.
At this point, you might note that this is all well and good, but what can be done with this information? An accompanying editorial in Nature lets us know what must come next if any of this is to benefit patients. After noting that when cancer genome sequencing projects were first launched the hope was that they would result in a catalog of mutations that can give rise to a cancer and that this core mission has now been achieved, the editorial notes:
Cancer genomes have been sequenced for more than a decade, but now researchers and the funders who support them must tackle the next challenge. The goal has always been to improve the lives of those affected by cancer, and the reams of data amassed by sequencing projects have helped. They are used by researchers to find new drug targets, and to generate new markers that can be used to match patients with the treatment most likely to help.
But most of the data so far have been limited in one crucial respect: clinical details of the sample donors are often missing. The first samples collected for the Cancer Genome Atlas, a sequencing project that ran from 2006 to 2018, co-funded by the US National Cancer Institute and the National Human Genome Research Institute, typically came with little more than the donor’s gender, diagnosis and age at diagnosis. Rarely would there be a record of that person’s family or medical history, what therapy they had received and how they had responded — all crucial information if genome sequences are to be put to work to help patients.
Yes, we have all this information now, but what we also know is that, as I like to describe while channeling Douglas Adams, cancer is complicated. You just won’t believe how vastly, hugely, mind-bogglingly complicated it is. I mean, you may think your income tax return is complicated, but that’s just peanuts compared to cancer.
In any event, to reiterate things I’ve written about in the past, the vast majority of cancer-causing driver mutations can be divided into a relatively few key molecular signaling pathways. As these most recent papers found, confirming previous findings, mutations at the “trunk” of the divergent branching evolution tend to remain as divergent evolution occurs, which is why targeting them is more likely to be effective than targeting mutations further out on the “branches.” Unfortunately, what this study also shows is that, thanks to evolution, curing a given cancer is just not going to be as simple as getting a biopsy of a tumor and picking a targeted agent or two to blast the tumor into oblivion. Not only is cancer not one disease, even a given cancer is in a given patient is arguably not just one disease but many as evolution leads its cells to become more and more heterogeneous. This doesn’t mean that true “personalized medicine” or “precision medicine” is impossible or that it’s impossible to develop cures for various cancers. The point is just that, as this study shows us yet again, that biology is always way more complicated than we ever thought it was, and evolution almost always wins out in the end.