
{kind=link}
[Note: In the absence of a new post today, here’s an updated, old post, that you may not have seen.]
With healthcare costs continuing to rise, generic drugs are looking more attractive than ever. The prospect of getting the same drug at a lower cost is tempting to anyone with a large drug bill — patient or insurer alike. The savings to insurers and individuals are massive: prices can drop by 90% or more, almost overnight. In 2012, Plavix and Seroquel, two other blockbusters, lost patent protection too — with just two drugs, it was estimated there would be $10 billion in health care savings created. A “patent cliff” that existed several years ago was expected to shrivel about $255 billion in worldwide patented drug sales over the following five years. If you’re taking a prescription drug and not already on a generic version, you may be in the future.
Pharmacists are responsible for most of the prescription switches from brand to generic drugs. It depends on the state or province, but in some cases generics may be automatically substituted — that is, switched without patient or prescriber consent. Misconceptions are common, ranging from manufacturing standards (“lax standards!”) to efficacy (“the drugs don’t work!”). So today’s post is an overview of the science of evaluating generic drugs. Specifically, I want to review the concept of bioequivalence, the confirmation of which assures us of the interchangeability of different drugs — that is, one can be substituted for another.
What is a generic drug?
What is referred to as a “generic” drug may vary by country, and be influenced by both medical practice and by regulatory requirements. The most common definition is that used by the World Health Organization:
A generic drug is a pharmaceutical product, usually intended to be interchangeable with an innovator product, that is manufactured without a licence from the innovator company and marketed after the expiry date of the patent or other exclusive rights.
Generic products may also be called “multisource” products. And you’ll often see a drug’s chemical name referred to as the “generic name” or the “non-proprietary” name, which as I’ve described can lead to confusion among consumers who may only know their prescription by the brand name alone.
The active pharmaceutical ingredient
To understand the scientific basis of generic drug evaluations, it’s necessary to understand some key concepts. The first one is the active pharmaceutical ingredient or API. In Lipitor, for example, the API is atorvastatin, or to use its full chemical name, (3R,5R)-7-[2-(4-fluorophenyl)-3-phenyl-4-(phenylcarbamoyl)-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate. The API is the chemical that has the desired biological effect. There may be a dozen ingredients in a tablet, for example, but the API is the ingredient we’re interested in. It’s the API that will allow us to generalize data and studies with the drug, linking the original bench science and preclinical research, to the tablet dispensed by the pharmacy — it’s the same chemical. The fact that drugs have an API allows generic drugs to be marketed, because when we compare generics, the API is the same. In contrast, consider the scenario of an herbal remedy. A single tablet of 100mg of a raw herb might contain hundreds of different chemicals. If there is no known API or standardized active ingredient, we cannot compare between brands, or assume that clinical trials with one brand are relevant to any other product, because we have no idea which ingredient is actually having an effect, and if any other version has that same ingredient (or combination of ingredients.) That’s why understanding the active components in herbal medicines is so important.
Bioavailability
Most dosage forms (e.g., tablets and capsules) are designed to deliver the API to the site of action. Unless it’s a drug that acts directly on the lining of the gastrointestinal tract, we rely on the circulatory system to carry the drug to the site of action in the body. Bioavailability refers to the amount of drug that, once ingested, reaches the bloodstream. Bioavailability is evaluated based on two measures — the rate of absorption, and the extent of absorption:
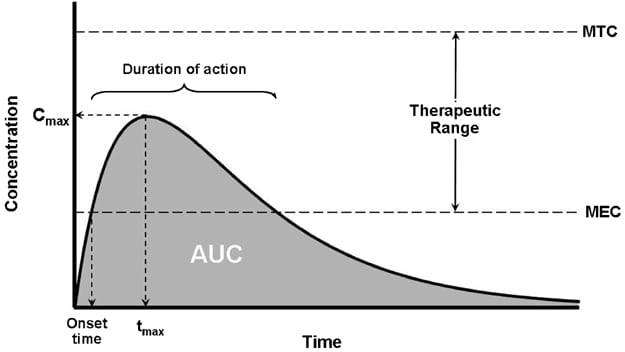
Look at this graph carefully. Let’s assume a dose of a drug is taken by mouth at time=0. A dosage form, such as a tablet, needs to disintegrate, and then the API needs to dissolve. It then must be absorbed through the gastrointestinal tract, where it will move into in the blood. Sequential blood samples are taken (say, every 30 minutes) and measured for the concentration of the drug. The point measurements are then connected, resulting in the smooth line you see. At first, the drug is absorbed rapidly, peaking at concentration Cmax at time tmax. The drug then starts to be eliminated from the blood — it could be metabolized, perhaps by the liver, and/or be filtered out of the circulation, by the kidneys. We’re then able to calculate the rate of absorption, described by Cmax/tmax, and the total extent of absorption and exposure in the body, which is the area under the curve (AUC).
Studying the physiological actions of the API, and relating it to the concentration time profile allows us to understand if there is a maximum tolerated concentration (MTC) and if there’s a certain blood concentration where the drug’s action seems to start and wear off, termed the minimum effective concentration (MEC). These values are estimates, based on the understanding that there is a relationship between the drug’s concentration in the bloodstream, and the amount of that same drug at the site of action in the body. These measures will guide how the drug is eventually manufactured: We want each dose to predictably and reliably follow the same curve. That means standard production methods, and the use of fillers, diluents and other excipients, all of which serve to ensure that there is little to no variation tablet-to-tablet or dose-to-dose. This is done with the intention of also minimizing interpatient variability, which is the variation seen between different patients given the same dosage form.
There may be other ways that the formulation can be varied. When the MEC is very close to the MTC, greater precision in per-tablet accuracy may be necessary. Further, we can vary shape of the curve, by doing things like enteric-coating the tablet, or making a sustained-release version that releases the API more slowly. The final absorption curve of any pharmaceutical preparation will be a product of both the physical characteristics of the drug (i.e., its intrinsic chemical properties) and the dosage form (i.e., the pharmaceutical properties).
Generic manufacturers that want to duplicate a drug coming off patent then have a few challenges. First, manufacture the complicate chemical structure that is the API (or find someone else who can make it for you). Second, package it in a dosage form that resembles the patented drug. Third, show that the new generic shares the same absorption curve as the branded version. That is, they must show that two product are bioequivalent.
Bioequivalence
We now want to compare a branded drug, one that’s been on the market for years, with a new generic. There is a single fundamental assumption that underlies the comparison:
Two products are considered equivalent when the rate and extent of absorption of the generic drug does not show a significant different from the rate and extent of absorption of the brand drug, when administered at the same dose under similar experimental conditions.
So let’s unpack this. We don’t need to know if the tablets look alike, or have different fillers. We also don’t need to do clinical trials with the generic drug. If we can demonstrate that the API is absorbed at the same rate and extent as the brand drug, then we can declare two products to be bioequivalent. That is, taking one or the other results in the same “body exposure” to the same drug. Assume we want to compare Drug A and Drug B. We will take a group of normal, healthy adults and put them in standardized conditions — usually fasting overnight and then giving a drug on an empty stomach or with a standardized meal. We’ll give Drug A to a patient, take serial blood samples, and then calculate Cmax/tmax and AUC. Later, after the drug has been fully eliminated from the body, we’ll repeat the process in the same patient, under the same conditions, with drug B. This may be done in 20+ patients. We’ll get a graph that looks like this:
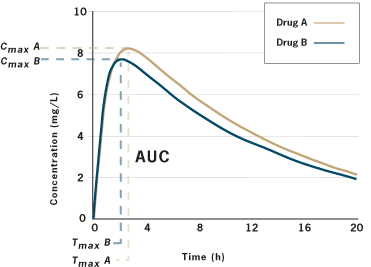
Typical concentration-time graph for two drug products
(source)
Are the two curves super-imposable? Not quite, in this example. There are slight differences in the extent of absorption. Are these two drugs bioequivalent? Here’s where the statistics come in. We apply statistics to evaluate if the curves are meaningfully different. To do this, we need to make some assumptions about what we will accept as a meaningful difference. Does a slight difference in the rate or extent of absorption make a clinical difference? Most regulators worldwide have decided that a 20% variation is generally not clinically significant.
Two versions of a drug are generally said to be bioequivalent if the 90% confidence intervals for the ratios of the geometric means (brand vs. generic) of the AUC and Cmax fall within 80% and 125%. The tmax (brand vs. generic) must also be comparable — and there should not be any significant differences between different patients.
For practical purposes, generic versions of branded drugs have AUC and Cmax ratios that are very close to one. With significant variation in either value, it would be unlikely for the confidence intervals to lie within the 80% to 125% range. For the sake of keeping this post short I’ll leave a more detailed discussion of the statistics to the interested reader.
Myths, misconceptions, and controversies
Generic drugs are manufactured differently from branded drugs, and branded drug manufacturers use better processes
If the dosage form releases the same drug with the same concentration/time profile, then any minor manufacturing differences are irrelevant to the pharmacological activity. Regulators have established standardized manufacturing practices and standards, termed good manufacturing practices, that all manufacturers, brand or generic, must adhere to. Final products must meet the same product quality standards as well.
Biosimilars aren’t generics
Biosimilars refers to follow-on versions of antibodies, proteins and other patented biological drugs, like growth hormones. Biological drugs are very large molecules that are structurally more complex that the relatively simple, smaller molecules in most tablets and injections. Their complexity means that a simple bioequivalence evaluations may be insufficient to fully describe a drug’s action. Manufacturing processes may involve recombinant DNA technology, and can take place inside living cells, leading to a final dosage form which cannot be fully characterized as a single defined API. As some of the biologic drugs approved over the past few decades have lost patent protection, the act of verifying both pharmaceutical but also clinical equivalence of “generic” biologicals has become an evaluation challenge for regulators. So if they’re not completely bioequivalent, do they have the same effects in patients? That is, are they clinically and therapeutically equivalent? While approaches between countries may vary, many are being cautious and requiring more detailed comparisons, including clinical safety and efficacy trials to demonstrate that biosimilars have the same end effects. As regulators gain more experience evaluating these products, expect the testing standards to evolve. (There’s good information for health professionals and consumers prepared by the Canadian Agency for Drugs and Technologies in Health which provides more detail on the current evaluation process.)
Bioequivalence studies are always necessary to establish interchangeability
For some dosage forms, bioequivalence studies may be inappropriate or impractical. Consider ophthalmic eyedrops. These products are administered at the site of action, and don’t rely on systemic absorption. It may be more appropriate to verify equivalence by comparing physicochemical properties including pH, viscosity, and osmolarity/osmolality. Where all relevant parameters are evaluated to be comparable, regulators will deem generic versions interchangeable based on these parameters alone.
Generics need to be tested in sick patients, not healthy volunteers
The intent of a bioequivalence comparison is to determine the differences in formulation between two drug products. Healthy standardized volunteers are used in bioequivalence studies to control for patient differences, meaning that any differences in bioavailability will be the result of drug formulation issues, not patient issues. In order to isolate drug formulation effects, it is necessary to hold everything else as constant as possible. If formulations are consistent, the rate and extent of drug absorption will not differ, and consequently there should be no differences in the pharmacologic effects of the drugs.
I absolutely can’t take a generic version of my prescription
Patented drug manufacturers usually aren’t willing to sacrifice their entire market share to generic competitors. Some will start producing “generic” versions of their own drug, selling them to a partner or subsidiary who will sell them as an “ultrageneric”. So in many cases, at least one of the generic versions that’s marketed will be truly identical (except perhaps for markings) to the branded products. Now if you think this means people won’t occasionally identify the switch as the source of efficacy issues or side effects, my anecdotal experience says otherwise.
I had a negative reaction (e.g., more side effects) to the generic
There are no requirements for generic drugs to contain the same non-medicinal ingredients as the brand-name drug, so allergic reactions to generic versions of drugs are possible. Most of the excipients are generally inert but there have been suggestions that serious allergies may rarely affect some patients. However, complaints about intolerance in the absence of a true allergic response or documented allergy could also be nocebo effects, negative symptoms elicited by negative expectations from the patient.
Bioequivalence testing isn’t an accepted evaluation method
It’s not just generic companies that must do bioequivalence studies. The company that owns the patent may need to do the same studies. Any changes to the dosage form either during development, or after marketing the product, may necessitate bioequivalence studies to demonstrate that changes do not meaningfully alter the concentration-time curve. So if the clinical trial was with a capsule, and the final marketed product is a tablet, a bioequivalence study comparing the two may be necessary. This reassures regulators that the clinical trials continue to be relevant and applicable to the new dosage form.
I don’t care what you say, the drugs are not equivalent in the real world
The Medical Letter summarized some studies that have evaluated the clinical effects of generic substitution:
- A systematic review of 47 studies that compared cardiovascular drugs found no evidence that brand-name drugs were superior with respect to clinical outcomes.
- An RCT comparing a brand antibiotic vs. a generic antibiotic found no differences in clinical effects.
- The FDA conducted its own evaluation of proton pump inhibitors (e.g., Prilosec/Losec). All five generics met dissolution standards.
- Levothyroxine as an API is unstable, and the FDA recently tightened manufacturing standards for all versions. There are no well-documented cases of therapeutic inequivalence between brands deemed to be bioequivalent. Most health professionals still recommend using a consistent brand, however.
- Retrospective case-control studies with anti-epileptic drugs have yielded some data suggestive of therapeutic inequivalence. Given the limits of these studies (patients who have switched brands may differ in some way from those that did not switch), the data are not conclusive. Clinically, most health professionals are cautious when switching to generic antiepileptic drugs.
Conclusion: Bioequivalence testing ensures safe, effective generics
The science of bioequivalence evaluations for generics has been in place in most countries for more than 20 years with an established track record of therapeutic equivalence. These evaluation methods have been so successful in establishing generic drug standards that they are largely consistent between all of the major drug regulators worldwide. Consumers and health professionals alike can be reassured that generic drugs approved under these regulatory frameworks are indeed bioequivalent, and therefore, interchangeable with brand name products.
Photos via flickr user Chris Potter (ccPixs.com) used under a CC licence.