{kind=link}
One of the more difficult conversations to have with a patient as a cancer doctor occurs when a patient, recently informed of her diagnosis of, for example, breast cancer, asks me, “Why did I get this? What caused it?” What almost inevitably follows is an uncomfortable conversation in which explanations of the multiple known causes of breast cancer do not satisfy the patient. The reason, of course, is because when a patient asks, “What caused it?” she doesn’t mean what causes breast cancer in general or in statistical terms. Rather, she means, what caused my breast cancer? It’s a question that can only occasionally be answered. For instance, if it’s lung cancer and the patient is a smoker, then it was almost certainly smoking that caused the cancer, because lung cancer is a relatively rare cancer in the absence of smoking. In the case of breast cancer, contrary to the prevailing belief that leads women with breast cancer to be puzzled about how they could get it when there’s “no cancer” in their families, only around 5-10% of cases have a familial or genetic component. That means that around 90% of breast cancers are what we call “sporadic,” which means that we can’t identify a specific cause. Or, as I like to say, “We just don’t know.” Worse, in the case of breast cancer, the environmental factors we know about appear to contribute modestly at best to the risk of cancer. (More on this later.)
Understandably, patients hate hearing “We just don’t know,” some vague handwaving about genes, and that there is nothing that we know of that they did that caused their cancer. People—including oncologists—really don’t like the concept of “sporadic” cancer, mainly because humans crave explanation. The default assumption is that everything must happen for a reason and there must be a cause for every disease or cancer. Perhaps the most ridiculously emphatic statement of this that I’ve encountered thus far comes from (who else?) über-quack Mike Adams when he heaped contempt on the idea of sporadic disease as “spontaneous disease.” He did this in the context of a story describing how, after Dr. Mehmet Oz had followed recommended care and undergone screening colonoscopy to look for polyps, he was shocked that he actually had some. This led Adams, in his usual inimitable fashion, to construct a straw man so massive that it could be seen from space when he set it on fire, declaring that “colon polyps, in other words, appear without any cause!” and that “mainstream medicine…believes in the theory of ‘spontaneous disease’ that ‘strikes’ people at random.”
Not exactly.
On the other hand, there is a lot of randomness in disease, not just cancer, as hard as it is for Mike Adams, or anyone to accept. Just because there is a varying amount of randomness in who gets a disease does not mean that mainstream medicine claims there is no cause to these diseases. Rather, for diseases like cancer, it’s a stochastic process, meaning that chance can play a role—sometimes a big role—in determining who gets sick. Indeed, just last week there was more evidence supporting this idea published in Science. Unfortunately, much of the mainstream press coverage presented the message of the paper a bit too simplistically. Even more unfortunately, it was the authors who encouraged this, as did the Johns Hopkins University press release about the study, which was entitled “Bad Luck of Random Mutations Plays Predominant Role in Cancer, Study Shows“. Yes, I groaned when I read this title.
Other headlines followed Hopkins’ lead:
- Math Suggests Most Cancers Are Caused By “Bad Luck”
- Two-thirds of cancers caused by bad luck, not heredity & environment
- Besides Lifestyle and Inherited Genes, Cancer Risk Also Tied to Bad Luck
- Biological bad luck blamed in two-thirds of cancer cases
I groaned. Then it was once more unto the breach.
Is cancer mostly “bad luck”?
The paper itself, by Cristian Tomasetti, a biostatistician interested in cancer evolution, genomics, and stem cell dynamics, and Bert Vogelstein, the latter of whom is a giant in cancer research, the man who pioneered the concept of tumor suppressor genes, validated p53 (TP53) as a tumor suppressor gene, discovered (with collaborators) the APC tumor suppressor gene, and whose team developed a major model explaining the progression of colorectal cancer from normal epithelium to polyp to dysplastic polyp to in situ cancer to cancer, is entitled “Variation in cancer risk among tissues can be explained by the number of stem cell divisions“. This, of course, is a different message than what is being promoted in the Hopkins press release and all the news stories that flowed from that. It is, however, a fascinating paper, although certainly not without issues, and it does support the idea that a large percentage of cancers tend to be due to mutations that occur during DNA replication during cell division.
The authors start out by noting:
Extreme variation in cancer incidence across different tissues is well known; for example, the lifetime risk of being diagnosed with cancer is 6.9% for lung, 1.08% for thyroid, 0.6% for brain and the rest of the nervous system, 0.003% for pelvic bone and 0.00072% for laryngeal cartilage (1–3). Some of these differences are associated with well-known risk factors such as smoking, alcohol use, ultraviolet light, or human papilloma virus (HPV) (4, 5), but this applies only to specific populations exposed to potent mutagens or viruses. And such exposures cannot explain why cancer risk in tissues within the alimentary tract can differ by as much as a factor of 24 [esophagus (0.51%), large intestine (4.82%), small intestine (0.20%), and stomach (0.86%)] (3). Moreover, cancers of the small intestinal epithelium are three times less common than brain tumors (3), even though small intestinal epithelial cells are exposed to much higher levels of environmental mutagens than are cells within the brain, which are protected by the blood-brain barrier.
Another well-studied contributor to cancer is inherited genetic variation. However, only 5 to 10% of cancers have a heritable component (6–8), and even when hereditary factors in predisposed individuals can be identified, the way in which these factors contribute to differences in cancer incidences among different organs is obscure.
So this is what we know about cancer. Its incidence varies tremendously among tissues in a way that can’t be explained by environmental exposure or heredity. I might quibble with the authors’ not discussing lung cancer more as a major exception to this tendency, given that nearly all lung cancer is caused by tobacco smoking, but overall for most other cancers this is basically accurate. Given the inadequacy of environmental factors and inherited genetic mutations as explanations for how cancers arise, the authors then tell the reader how this led them to look at what they call a “third factor”: the stochastic effects associated with the lifetime number of stem cell divisions within each tissue. The first thing that struck me as I read this paper is that this is not a new or radical idea. Far from it! It’s long been known that tissues with cell types that undergo rapid division over a lifetime tend to be more susceptible to developing cancer; e.g., the epithelial cells of the colon, which are prone to cancer, compared to cancers arising in the brain or in muscle, where cells divide much less. The reason, it’s been hypothesized, is that more rapid cell division leads to more errors in DNA replication, which leads to more mutations. However, there are glaring exceptions, such as the small intestine, whose epithelial cells replicate rapidly and continuously throughout the human lifespan – yet small bowel cancer is rare.
The question most readers have, probably, is why Tomasetti and Vogelstein wanted to concentrate on stem cells. The reason is simple and fairly obvious. Most cells are fully or partially differentiated, and a relatively small proportion of cells in most tissues and organs are responsible for the development and maintenance of the tissue and its architecture, as well as for replacing cells that are lost. As is pointed out in the paper, until recently, the nature, number, and hierarchical division patterns of various stem cells were not well characterized. In any case, the hypothesis being tested in this paper relies on three key concepts: (1) that cancer is mostly the result of acquired genetic and epigenetic changes; (2) genetic changes occur primarily by chance during DNA replication; and (3) that the endogenous mutation rate of all human cell types appears to be very similar. These concepts, taken together, predict that the number of cell divisions among stem cells in each organ during a lifetime should correlate strongly with the lifetime risk of cancer in that organ. To test this prediction, Tomasetti and Vogelstein set out to estimate the lifetime number of stem cell divisions in each organ and determine if it correlates with cancer risk in that organ.
So Tomasetti and Vogelstein reviewed the current literature, identifying 31 tissue types in which stem cells had been quantitatively assessed. They then plotted the total number of stem cell divisions during the average human lifespan on the X-axis and the lifetime risk for cancer as recorded in the Surveillance, Epidemiology, and End Results (SEER) database in that organ on the Y-axis. Here’s the end result in Figure 1:
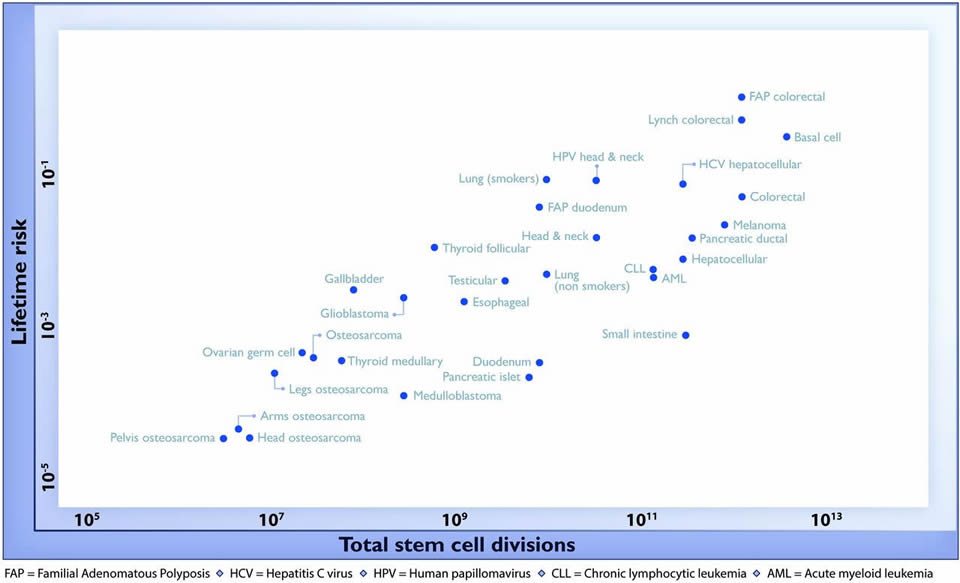
As you can see, there is a striking, highly-positive correlation on a log-scale graph. As noted by the authors, this correlation extended across five orders of magnitude and encompassed cancers with tremendous differences in prevalence. The authors note:
No other environmental or inherited factors are known to be correlated in this way across tumor types. Moreover, these correlations were extremely robust; when the parameters used to construct Fig. 1 were varied over a broad range of plausible values, the tight correlation remained intact…
A linear correlation equal to 0.804 suggests that 65% (39% to 81%; 95% CI) of the differences in cancer risk among different tissues can be explained by the total number of stem cell divisions in those tissues. Thus, the stochastic effects of DNA replication appear to be the major contributor to cancer in humans.
It seems pretty bullet-proof, right? Well, maybe. Maybe not. As was pointed out by Paul Knoepfler, the single most difficult issue in this analysis is how to accurately and consistently calculate reliable estimates of the number of lifetime stem cell divisions in a series of diverse tissues. This is far from a trivial matter, or, as Dr. Knoepfler describes it, “extremely difficult and it’s not difficult to imagine such calculations being off by one or more orders of magnitude.” For instance, take a look at this description in the supplemental data of how stem cell divisions were calculated for adenocarcinomas and squamous cell carcinomas of the esophagus (which I’ll quote because I realize that most of you don’t have access to the paper itself):
The lifetime incidence of cancer of the esophagus is ~0.51% (3). The vast majority of these cancers are either adenocarcinomas or squamous cell carcinomas, and the ratio of these two types has changed considerably in the last few decades. Currently, ~38% of esophageal cancers are squamous cell carcinomas (66, 67). The lifetime incidence of esophageal squamous cell carcinoma is therefore 0.0051•0.38 = 0.001938. The esophagus is 18 to 26 cm long and 2 to 3 cm in diameter (68), resulting in an average surface area of ~172.79 cm2. The area of a squamous cell in the basal layer is ~80μm2 (69). The fraction of stem cells in the basal layer has been estimated to be 0.4% of the total basal layer cells (70), so there are a total of ~2.16•108 basal cells and 8.64•105 stem cells in the basal layer. As the normal mucosal epithelium is ~10-20 layers thick, the total number of epithelial cells in the esophagus is estimated to be ~2.16•108 •15= 3.24•109. Cells have been estimated to turn over ~ every 21 days (71, 72).
As you can see, multiple assumptions had to be made for each parameter, and then these parameters are multiplied together to estimate the total lifetime number of stem cell divisions. To be fair, one thing Knoepfler didn’t mention is that there was a statistical analysis done to test the robustness of the correlation in which the estimates of total stem cell divisions was allowed to vary by 100-fold in either direction, but even so, there are a number of other issues. For instance, not all cancers originate with stem cells, and some organs have more than one type of stem cell population. Yet the paper seemed to treat stem cells as one population for each organ. Still, as a first approximation, it’s not unreasonable to do it this way.
In addition, to try to tease out which tissues are more prone to cancer than their stem cell replication totals would suggest, Tomasetti and Vogelstein calculated what they call an “extra risk score” (ERS), defining it as the product of the lifetime risk and the total number of stem cell divisions (in log10). Machine learning methods were then used to classify tumors based only on this score. The details are a bit much, even for me, but the bottom line is that the ERS is meant to be a measure of how well that particular tumor type “fits the model.” More specifically:
If the ERS for a tissue type is high—that is, if there is a high cancer risk of that tissue type relative to its number of stem cell divisions—then one would expect that environmental or inherited factors would play a relatively more important role in that cancer’s risk (see the supplementary materials for a detailed explanation). It was therefore notable that the tumors with relatively high ERS were those with known links to specific environmental or hereditary risk factors.
The authors referred to tumors with high ERS values as “D-tumors” (for “deterministic,” meaning deterministic factors such as environmental mutagens or hereditary predispositions strongly affect their risk) and tumors with low ERS values as “R-tumors” (for “replicative”) in which stochastic factors, most likely errors during DNA replication as stem cells divide, most strongly affect risk. Not surprisingly, cancers that came up as D-tumors with high ERS scores included colorectal cancer related to familial adenomatous polyposis (FAP), which is due to a mutation in a tumor suppressor gene (APC) and whose most severe form results in a 90+% risk of colorectal cancer by age 50, and to Lynch syndrome (HNPCC or hereditary nonpolyposis colorectal cancer), which is due to an autosomal dominant inherited mutation that produces a high risk of colorectal cancer, as well as cancers of the endometrium, ovaries, stomach, small intestine, hepatobiliary tract, upper urinary tract, brain, and skin. Lynch syndrome is due to defects in DNA mismatch repair, which lead to—you guessed it—more mistakes during DNA replication, leading to more mutations. People with Lynch syndrome have an 80% lifetime risk of colorectal cancer. Indeed, in general, so high is the risk of colorectal cancer in people with FAP or HNPCC that close monitoring with regular colonoscopies is always recommended, and prophylactic surgery to reduce the risk of cancer by eliminating the tissue at risk is frequently recommended. The recommended operation is usually total proctocolectomy (removal of the entire colon and rectum) as young adults, although because there are less aggressive variants of FAP sometimes close monitoring is considered more appropriate.
One little nit I have to pick with this graph is that the lung cancer cases are divided into smokers and non-smokers. Again, lung cancer, which is such a leading cause of preventable cancer (with the authors noting that smoking increases lung cancer risk 23-fold in men and 13-fold in women, for an overall 18-fold increase in risk), is a special case, but that doesn’t justify dividing the cases of lung cancer into those related to smoking and those unrelated to smoking. Doing so smacks of trying to downplay one case of environmental factors being very important. After all, there are other cancers whose incidence is very much influenced by smoking, including esophageal cancer, head and neck squamous cell cancers, and pancreatic cancer, but no such effort was made to divide these tumors into smoking-induced and not smoking-induced. This lapse is curious and not explained in the paper at all. Either all the strongly smoking-induced cancers or none of them should have been divided into groups based on smoking.
Another issue is that most of the “D-tumors,” the ones known to have definite genetic or environmental causes that fell out were tumors caused by genetics. Of tumors known to have a strong environmental component, only smoking-induced lung cancer, basal cell carcinoma (due to sun exposure), and thyroid cancer (follicular or papillary types, known to be significantly due to radiation exposure) showed up in the D-tumors group. This made me wonder if Tomasetti and Vogelstein’s model was sensitive enough to pick up tumors that have a significant environmental component, given that environmental influences tend not to produce cancer risks as high as those seen in people harboring genes for inherited cancers, for which mutant gene carriers can have up to a 90% lifetime risk of cancer when they have a particularly nasty genetic predisposition.
There are other problems. The first is a rather glaring anomaly. Oddly, malignant melanoma, which is known to be strongly associated with sun exposure, showed up as an “R-tumor,” and no discussion was offered to explain why this might have happened. Again, this brings into question the sensitivity of the model. Next, two major cancers, breast and prostate cancer, were not included in the analysis because Tomasetti and Vogelstein could not find good estimates in the literature for stem cell proliferation rates for these cancers. Given that these are two of the “big three” cancers with the highest incidence in the US (breast, prostate, and lung cancers), this omission presents a problem for generalizability. Overall, I wonder why Tomasetti and Vogelstein didn’t just show the first graph, which pretty much gives us all the information we need. The second analysis involved a transformation of the data whose rationale is not clear and even questionable, and that is not particularly informative. I understand why they did it, but I think it weakens the paper.
As has been pointed out elsewhere by Bob O’Hara and Grrlscientist, what is being represented as “bad luck” might or might not be that. What Tomasetti and Vogelstein’s calculations show is that, in their model, 65% of the variability in cancer incidence between tissues is attributable to variability in stem cell proliferation in those tissues, which, they argue, is not the same thing as what proportion of cancer is due to “bad luck.” What they are, in fact, describing is the stochastic component related to differences in stem cell proliferation, but “bad luck” is pithier and easier to understand. Therein, I suspect, lays their problem. It would have been a lot less interesting to the press to say that 65% of the variation in incidence between 31 cancers is explained by variations in lifetime stem cell proliferation in those tissues.
Perhaps the most reassuring thing about this paper, oddly enough, is that even after their having estimated the variability in incidence explained by stem cell proliferation and determined that 65% of tumors risk is correlated with stem cell proliferation and thus the accumulation of errors in replication, Tomasetti and Vogelstein’s estimate isn’t too far off from what we already know, as you will see. That’s part of the reason why I tend consider objections to the paper by bloggers like Bob O’Hara and GrrlScientist and The Stats Guy to be at least partially missing the point, at least from my perspective as a cancer biologist and clinician, in that they tend to focus on semantics (namely the use of the term “bad luck”) and purely statistical issues rather than considering these findings in context of what we already know about cancer biology. PZ Myers noted this problem before I did (damn this publication schedule in which I had to wait until Monday to post this!), but, for example, one of O’Hara and GrrlScientist’s key objections is:
So, what proportion of cancers are due to bad luck? Unfortunately it’s difficult to tell from the paper. The figure from the paper is on the log scale, and if we extrapolate the model to zero (no cell divisions), we’d see it assumes there is no risk of cancer.
To which PZ replied:
That’s what we’d expect if the magnitude of Mystery Factor X was 0, and the baseline rate of somatic errors leading to cancer was set by the replication error rate.
I’ll also add that this is not at all an unreasonable assumption as a first approximation for such a model. After all, without cell division there can’t be cancer, regardless of the source of cancer-causing mutations, which are random during DNA replication, epigenetic changes, or other sources. If the cells don’t divide, even mutations caused by something other than errors in DNA replication won’t be passed on and can’t accumulate to result in carcinogenesis. In any case, what Tomasetti and Vogelstein were correlating was the variability in cancer incidence that could be attributed to variations in stem cell proliferation. That’s it. Absolute cancer risk for each tissue isn’t the issue, but rather variability in cancer risk and what contributes to it.
In the ballpark
Let’s try to put Tomasetti and Vogelstein’s finding in context with respect to what we already know about cancer biology and epidemiology.
Any cancer biologist (like me), oncologist, or oncologic surgeon (like me) should know that we already have copious data for a large number of tumors that provide us with estimates of the relative effects of lifestyle and environment versus genetics versus randomness on the risk of developing cancer. Part of what bothered me about this paper is that this was not adequately discussed. (Actually, it was hardly discussed at all, but then, it is a Science paper, and Science imposes ridiculously tight word limits on most articles.) While it is not really possible to prevent most inherited cancers short of relatively radical or invasive interventions (chemopreventative drugs like Tamoxifen for familial breast cancer, which reduces the relative risk by only around 50%, or surgery to remove the organ before cancer can develop, as is done to prevent familial breast and colorectal cancer), it is theoretically possible to prevent a lot of cancer. I say “theoretically” because actually implementing the interventions designed to prevent these cancers would often be incredibly difficult as a practical matter. Just consider how difficult it has been to get people to quit smoking and lose weight if you don’t believe me.
Perhaps my favorite quick and dirty (not to mention, reliable) reference guide to estimates of what percentage of different cancers are potentially preventable is published on the Cancer Research UK website. On a page entitled Statistics on preventable cancers, you can peruse the estimated relative contribution to various factors, such as smoking, alcohol consumption, diet, sunlight, radiation, and others, to a wide array of different human cancers. Not surprisingly, by far the most common cause of preventable cancer is tobacco, causing an estimated 86% of cases of lung cancer, 65% of cases of esophageal cancer and cancers of the oropharynx and head and neck, 37% of cases of bladder cancer, and 29% of cases of pancreatic cancer. Diet is estimated to contribute to 31% of bowel cancers, 46% of esophageal cancer, 51% of stomach cancers, and 56% of head and neck cancers. Alcohol contributes to several cancers, including breast, bowel, esophageal, oral, and liver cancers, but the effects are much more modest. Sunlight contributes to 86% of cases of malignant melanoma, which, again, makes it very curious that melanoma showed up as an “R-tumor” in Tomasetti and Vogelstein’s analysis.
Overall, if you look at CRUK’s estimates of the percentage of cancers that are potentially preventable, you’ll find that, not surprisingly, it’s very high for smoking-related cancers like lung cancer (89%), esophageal cancer (75%), oral cancers (91%), pancreatic cancer (37%), and bladder cancer (42%). In contrast, it’s very low for cancers like non-Hodgkin’s lymphoma, prostate cancer, brain cancers, and myeloma. For the rest, it’s somewhere in between. Also included in the list are cancers caused by viral infections, such as cervical cancer, which can be prevented by preventing HPV infection.
One problem with much of the reporting of the study seems to be a misinterpretation of what the authors were getting at, which is stated near the end of the paper:
In formal terms, our analyses show only that there is some stochastic factor related to stem cell division that seems to play a major role in cancer risk. This situation is analogous to that of the classic studies of Nordling and of Armitage and Doll (10, 29). These investigators showed that the relationship between age and the incidence of cancer was exponential, suggesting that many cellular changes, or stages, were required for carcinogenesis. On the basis of research since that time, these events are now interpreted as somatic mutations. Similarly, we interpret the stochastic factor underlying the importance of stem cell divisions to be somatic mutations. This interpretation is buttressed by the large number of somatic mutations known to exist in cancer cells (14–16, 30).
In other words, even if taken at face value as reported in the media, Tomasetti and Vogelstein haven’t really demonstrated anything new. We’ve known for a long time that there is a strong stochastic (probabilistic) component to cancer development. The risk for some cancers appears to be mostly stochastic; the risk for other cancers is more strongly influenced by heredity; and the risk of other cancers, such as lung cancer, is more strongly influenced by environment. Some are influenced significantly by all three. These observations are, quite frankly, trivial. What Tomasetti and Vogelstein have done that’s different is to recast the stochastic component of cancer risk as not being due to just any old mutations in the cells in a tissue but rather to the accumulated mutations in stem cells, which is proportional to the total number of cell divisions. They then tried to estimate what percentage of cancers are primarily related to these stochastic factors and estimated that it’s around two thirds.
Now here’s the funny thing. If you interpret Vogelstein’s estimate the way it’s been represented in the press and by Vogelstein, as representing the percentage of cancers that are due primarily to “chance” and thus not related to environment and not potentially preventable (a not unreasonable interpretation, by the way), it turns out not to be such a bad first approximation at all. It’s actually not far off from what has been a fairly accepted estimate for quite a few years now, namely that between one third and one half of cancers are potentially preventable, implying that they are due to environmental causes that can be altered, such as smoking or diet. Given the level of uncertainty inherent in such estimates, even if you interpret Vogelstein and Tomasetti’s conclusion that two thirds of cancers are due to “bad luck,” their estimate of the percentage of cancers that are probably not preventable is definitely in the ballpark of commonly-accepted estimates, albeit at the lower end. Does that mean they’re on to something in concluding that stem cell replication over one’s lifetime primarily determines the “stochastic” component of cancer risk for each organ? That remains to be seen, but their preliminary finding makes sense, both from the perspective of producing a result that’s in the ballpark of what we already know based on epidemiology and being biologically plausible based on basic cancer biology.
There are two things that were probably responsible for the objections to this article. First was its being presented as concluding that two thirds of cancers are due to “bad luck.” While I understand that Vogelstein probably wanted to simply things so that they would be comprehensible to everyone, he might have simplified them a bit too much in search of the pithy, memorable phrase. (He certainly succeeded at getting the study noticed!) Lost in the shuffle was a more nuanced discussion of what Vogelstein was getting at when he said:
“All cancers are caused by a combination of bad luck, the environment and heredity, and we’ve created a model that may help quantify how much of these three factors contribute to cancer development,” says Bert Vogelstein, M.D., the Clayton Professor of Oncology at the Johns Hopkins University School of Medicine, co-director of the Ludwig Center at Johns Hopkins and an investigator at the Howard Hughes Medical Institute.
“Cancer-free longevity in people exposed to cancer-causing agents, such as tobacco, is often attributed to their ‘good genes,’ but the truth is that most of them simply had good luck,” adds Vogelstein, who cautions that poor lifestyles can add to the bad luck factor in the development of cancer.
That cancer is due to a combination of random probabilistic processes, environmental exposures, and heredity, is a non-controversial statement. What is controversial are estimates of the relative contribution of environment, given that the percentage of cancers due to inherited cancer-causing mutations is known and low. Take the example of breast cancer, which is a cancer for which environmental and lifestyle contributions are not particularly high, with perhaps 27% of breast cancers being due primarily to environment (which includes diet and exercise, as well as hormone replacement therapy). The vast majority of those environmental contributions come from obesity and alcohol consumption, neither of which reaches the double digits, percentage-wise. Yet there are organizations that promote the idea that “chemicals” in our environment are a major cause of breast cancer. Unfortunately, about 5-10% of breast cancer is inherited (which can only be prevented by aggressive means, such as chemoprevention or prophylactic surgery), while perhaps up to 27% has a strong environmental component. That leaves around 60% of breast cancer (or, even using higher estimates, at least 50%) as falling into the “we don’t know” or “stochastic” category, with, sadly, nothing that we know of right now that can be done to prevent these cases.
In other words, some of the objections to this paper seem to flow from a belief in inflated estimates of just what proportion of cancer is due to “environment” and is therefore potentially preventable. It’s been suggested that cancer biologists might be too fast to blame unknown causes on “randomness,” the assumption being that not knowing something means that we will know it in the future and more prevention will be possible. The problem is that not knowing something doesn’t mean that there’s a realistic way of obtaining that missing knowledge or that even if we obtained that knowledge that we’d be able to do anything with it. Also, from a biology standpoint, we already know a lot about the stochastic nature of cancer formation, and within this framework Tomasetti and Vogelstein’s study “makes sense,” although it might have under- or over-estimated the actual component due to errors in DNA replication in stem cells. Clearly the model needs refinement.
In any case, this sort of mindset showed up when, after Knoepfler’s analysis, a commenter named Catherine Danielson asked:
I have got to say that one question came to my mind instantly: who funded this cancer study? Sorry to be cynical, but when a study comes out that essentially clears all environmental factors from blame, I start to wonder if any of the funding came from, say, Monsanto. Whose interests might this study serve? Might we learn anything more if we follow the money? Maybe these questions have nothing to do with anything, but I think that they should be at least asked. We’ll certainly never find out the answers otherwise.
According to the acknowledgments, the work going into this paper, by the way, was supported by The Virginia and D. K. Ludwig Fund for Cancer Research, The Lustgarten Foundation for Pancreatic Cancer Research, The Sol Goldman Center for Pancreatic Cancer Research, and NIH grants P30-CA006973, R37-CA43460, RO1-CA57345, and P50-CA62924. No corporate funding there. It’s also a misstatement of the findings of the paper to say that it “essentially clears all environmental factors from blame”. You can argue whether it overestimates what percentage of cancer is related to variability in stem cell proliferation on some good statistical grounds, but a key finding of the paper is that at least one third of cancer incidence is not explained by this.
Unfortunately, Tomasetti and Vogelstein, did fall for another trap. Faced with their results, they demonstrated that they probably do not understand the concept of overdiagnosis and overtreatment:
The implications of their model range from altering public perception about cancer risk factors to the funding of cancer research, they say. “If two-thirds of cancer incidence across tissues is explained by random DNA mutations that occur when stem cells divide, then changing our lifestyle and habits will be a huge help in preventing certain cancers, but this may not be as effective for a variety of others,” says biomathematician Cristian Tomasetti, Ph.D., an assistant professor of oncology at the Johns Hopkins University School of Medicine and Bloomberg School of Public Health. “We should focus more resources on finding ways to detect such cancers at early, curable stages,” he adds.
Unfortunately, as I and others have documented here time and time again, early diagnosis is a lot more complicated an issue than even most physicians—even oncologists and radiologists—realize. Overdiagnosis and overtreatment are real phenomena because detecting cancers at ever-earlier stages and ever-smaller sizes picks up cancers that would never threaten the life of the patient, but we have to treat them as though they would because we can’t (yet) predict which ones are safe to watch and which ones will progress. Early diagnosis does not lead inevitably to improved survival. We’ve discussed this issue again and again here with respect to mammography, PSA testing for prostate cancer, screening for cancer in general, and screening for other diseases. More screening is not necessarily better.
In actuality, if Tomasetti and Vogelstein are correct, the proper conclusion would be to try to reduce environmental exposures for cancers known to have a large environmental contribution. For tumors that are largely driven by accumulated errors in DNA replication in stem cells we need to work on understanding the pathways that drive their growth and progression in order to develop better-targeted therapies, and to develop better surgical techniques to remove tumors for which extirpation is curative. Don’t get me wrong – early detection can be useful, but it is very cancer-type-specific and depends upon a tumor that progresses predictably from early pre-malignant stages and for which early intervention either prevents progression (as in removing polyps from the colon) or results in a better outcome (which is true of several cancers, but not to the extent that we like to think, being confounded by lead time and length biases). Finally, because screening sensitivity and specificity depend strongly on the prevalence of the cancer being screened for, screening only makes sense for the most prevalent cancers, like breast, prostate, lung, and colorectal.
It’s understandable that humans crave explanation, particularly when it comes to causes of a group of diseases as frightening, deadly, and devastating as cancer. In fact, both PZ Myers and David Colquhoun have expressed puzzlement over why there is so much resistance is to the concept that random chance plays a major role in cancer development, with Colquhoun going so far as to liken it to ” the attitude of creationists to evolution.” Their puzzlement most likely derives from the fact that they are not clinicians and don’t have to deal with patients, particularly given that, presumably, they do have a pretty good idea why creationists object to attributing evolution to random chance acted on by natural selection and other forces.
Clinicians could easily have predicted that a finding consistent with the conclusion that, as a whole, probably significantly less than half of human cancers are due to environmental causes that can be altered in order to prevent them would not be a popular message. Human beings don’t want to hear that cancer is an unfortunately unavoidable consequence of being made of cells that replicate their DNA imperfectly over the course of our entire lives. There’s an inherent hostility to any results that conclude anything other than that we can prevent most, if not all, cancers if only we understood enough about cancer and tried hard enough. Worse, in the alternative medicine world there’s a concept that we can basically prevent or cure anything through various means (particularly cancer), most recently through the manipulation of epigenetics. Unfortunately, although risk can be reduced for many cancers in which environmental influences can increase the error rate in DNA replication significantly, the risk of cancer can never be completely eliminated. Fortunately, we have actually been making progress against cancer, with cancer death rates having fallen 22% since 1991, due to combined efforts involving smoking cessation (prevention), better detection, and better treatment. Better understanding the contribution of stochastic processes and stem cell biology to carcinogenesis could potentially help us do even better.